

Immunotoxins and anticancer drug conjugate assemblies: the role of the linkage between components
- PMID: 22069744
- PMCID: PMC3202854
- DOI: 10.3390/toxins3070848
Immunotoxins and anticancer drug conjugate assemblies: the role of the linkage between components
Abstract
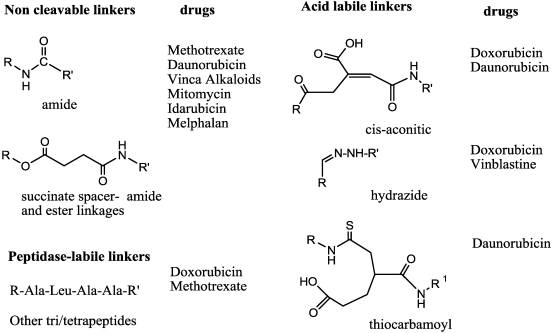
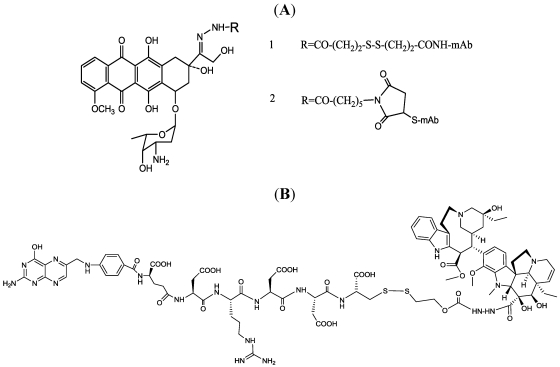
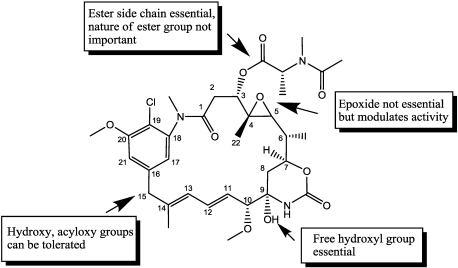
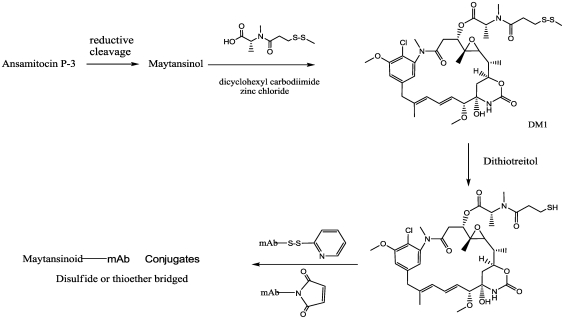
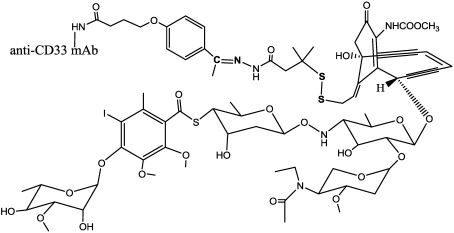
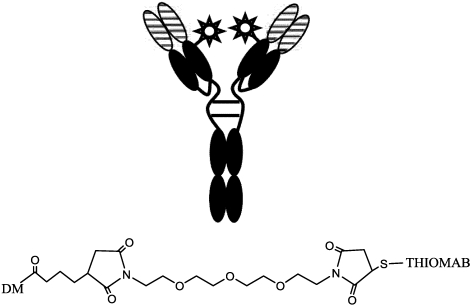
Immunotoxins and antibody-drug conjugates are protein-based drugs combining a target-specific binding domain with a cytotoxic domain. Such compounds are potentially therapeutic against diseases including cancer, and several clinical trials have shown encouraging results. Although the targeted elimination of malignant cells is an elegant concept, there are numerous practical challenges that limit conjugates' therapeutic use, including inefficient cellular uptake, low cytotoxicity, and off-target effects. During the preparation of immunoconjugates by chemical synthesis, the choice of the hinge component joining the two building blocks is of paramount importance: the conjugate must remain stable in vivo but must afford efficient release of the toxic moiety when the target is reached. Vast efforts have been made, and the present article reviews strategies employed in developing immunoconjugates, focusing on the evolution of chemical linkers.
Keywords: antibody drug conjugate; anticancer agents; conjugation process; immunotoxin; linker; toxins.
Figures
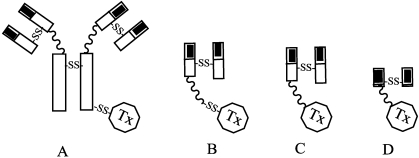
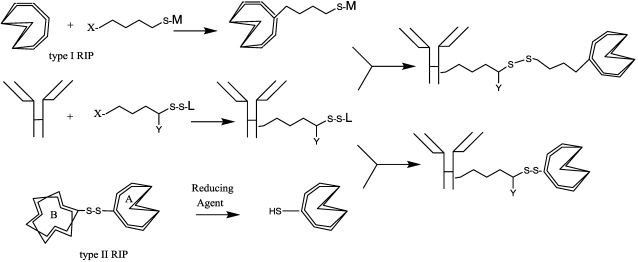
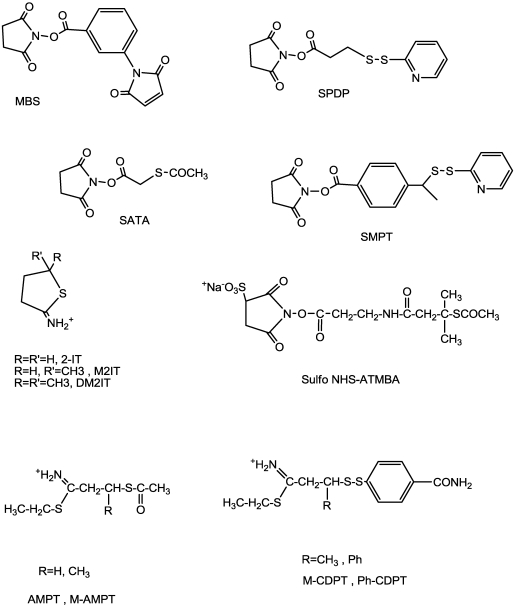
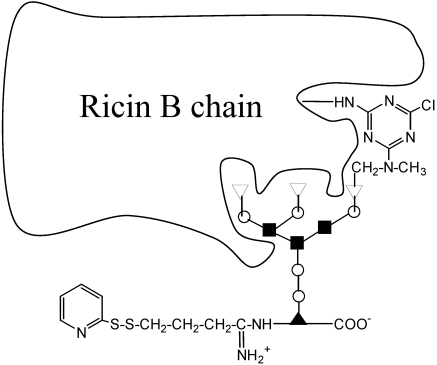
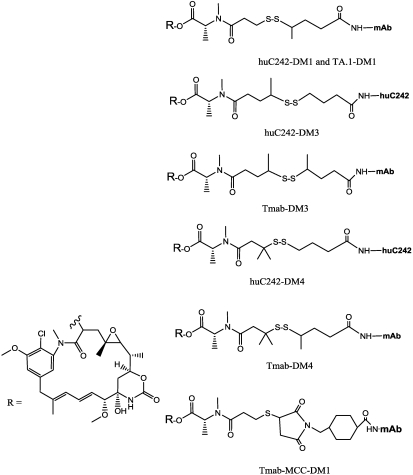
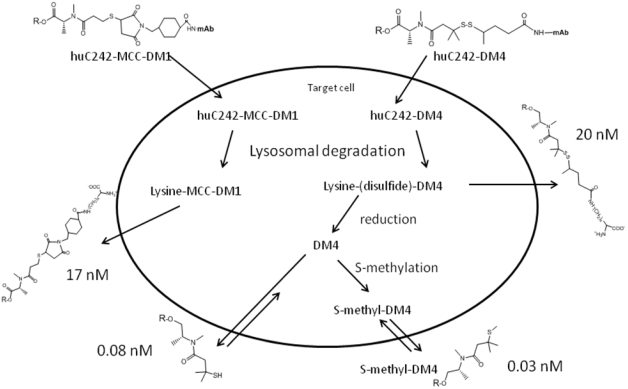
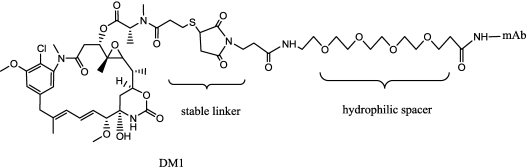
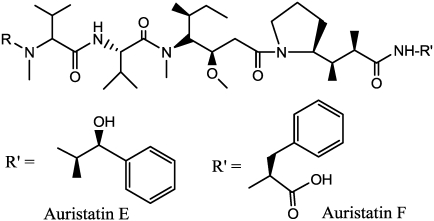
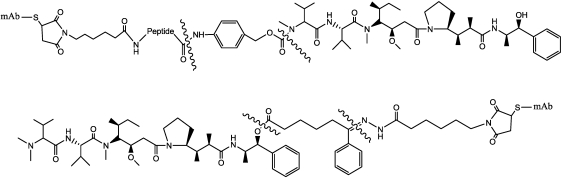
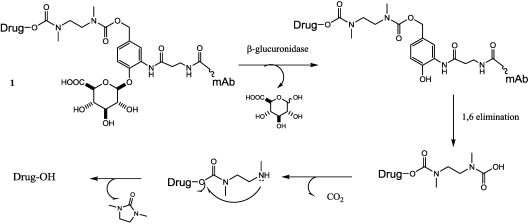
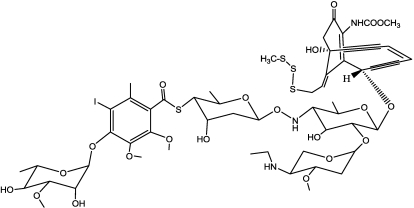
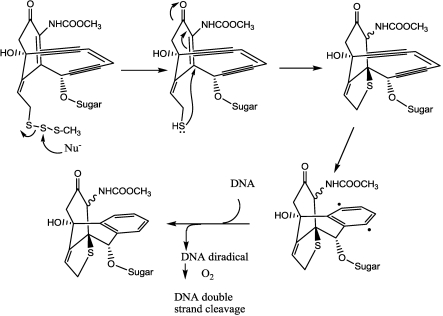
References
-
- Cancer facts and figures. [(accessed on 6 July 2011)]. Available online: http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/docume....
-
- Greish K., Fang J., Inutsuka T., Nagamitsu A., Maeda H. Macromolecular therapeutics: Advantages and prospects with special emphasis on solid tumour targeting. Clin. Pharmacokinet. 2003;42:1089–1105. - PubMed
-
- Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: Background and future prospects. Bioconjugate Chem. 2010;21:797–802. - PubMed
-
- Jain R.K. Transport of molecules across tumor vasculature. Cancer Metastasis Rev. 1987;6:559–593. - PubMed
-
- Roberts W.G., Palade G.E. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 1997;57:765–772. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
