

A critical window of CAG repeat-length correlates with phenotype severity in the R6/2 mouse model of Huntington's disease
- PMID: 22072510
- PMCID: PMC3349621
- DOI: 10.1152/jn.00762.2011
A critical window of CAG repeat-length correlates with phenotype severity in the R6/2 mouse model of Huntington's disease
Abstract
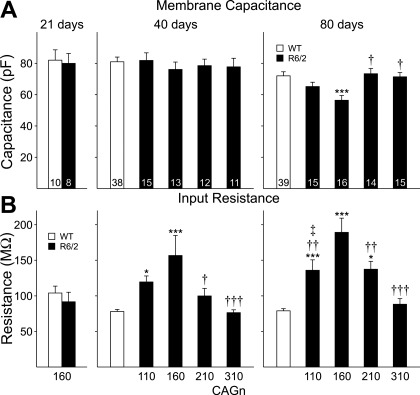
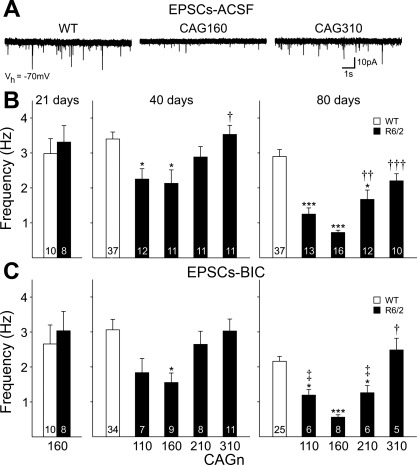
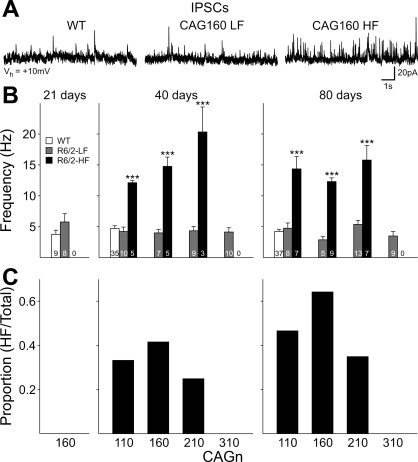
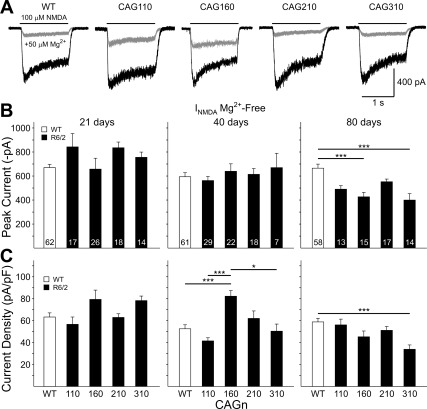
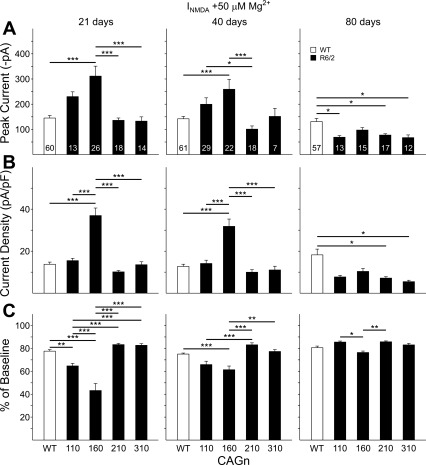
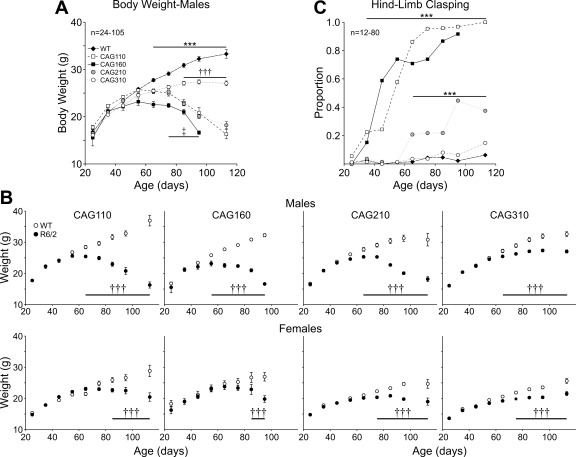
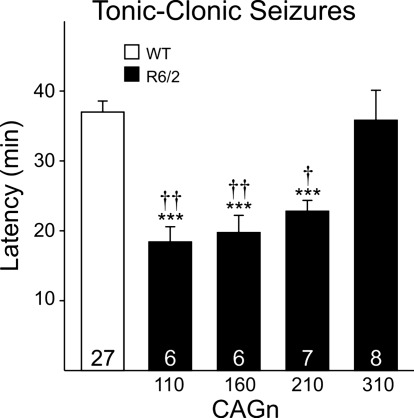
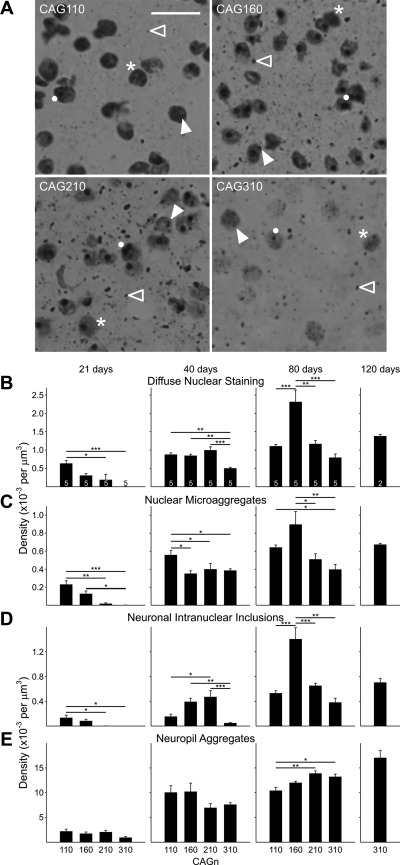
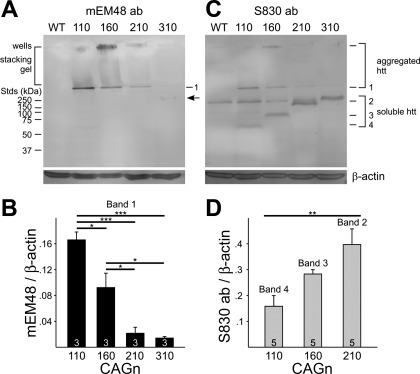
The R6/2 mouse is the most frequently used model for experimental and preclinical drug trials in Huntington's disease (HD). When the R6/2 mouse was first developed, it carried exon 1 of the huntingtin gene with ~150 cytosine-adenine-guanine (CAG) repeats. The model presented with a rapid and aggressive phenotype that shared many features with the human condition and was particularly similar to juvenile HD. However, instability in the CAG repeat length due to different breeding practices has led to both decreases and increases in average CAG repeat lengths among colonies. Given the inverse relationship in human HD between CAG repeat length and age at onset and to a degree, the direct relationship with severity of disease, we have investigated the effect of altered CAG repeat length. Four lines, carrying ~110, ~160, ~210, and ~310 CAG repeats, were examined using a battery of tests designed to assess the basic R6/2 phenotype. These included electrophysiological properties of striatal medium-sized spiny neurons, motor activity, inclusion formation, and protein expression. The results showed an unpredicted, inverted "U-shaped" relationship between CAG repeat length and phenotype; increasing the CAG repeat length from 110 to 160 exacerbated the R6/2 phenotype, whereas further increases to 210 and 310 CAG repeats greatly ameliorated the phenotype. These findings demonstrate that the expected relationship between CAG repeat length and disease severity observed in humans is lost in the R6/2 mouse model and highlight the importance of CAG repeat-length determination in preclinical drug trials that use this model.
Figures

References
-
- Alonso ME, Yescas P, Cisneros B, Martinez C, Silva G, Ochoa A, Montanez C. Analysis of the (CAG)n repeat causing Huntington's disease in a Mexican population. Clin Genet 51: 225–230, 1997 - PubMed
-
- Andresen JM, Gayan J, Djousse L, Roberts S, Brocklebank D, Cherny SS, Cardon LR, Gusella JF, MacDonald ME, Myers RH, Housman DE, Wexler NS. The relationship between CAG repeat length and age of onset differs for Huntington's disease patients with juvenile onset or adult onset. Ann Hum Genet 71: 295–301, 2007 - PubMed
-
- Ariano MA, Cepeda C, Calvert CR, Flores-Hernandez J, Hernandez-Echeagaray E, Klapstein GJ, Chandler SH, Aronin N, DiFiglia M, Levine MS. Striatal potassium channel dysfunction in Huntington's disease transgenic mice. J Neurophysiol 93: 2565–2574, 2005 - PubMed
-
- Bates GP, Hockly E. Experimental therapeutics in Huntington's disease: are models useful for therapeutic trials? Curr Opin Neurol 16: 465–470, 2003 - PubMed
-
- Benn CL, Landles C, Li H, Strand AD, Woodman B, Sathasivam K, Li SH, Ghazi-Noori S, Hockly E, Faruque SM, Cha JH, Sharpe PT, Olson JM, Li XJ, Bates GP. Contribution of nuclear and extranuclear PolyQ to neurological phenotypes in mouse models of Huntington's disease. Hum Mol Genet 14: 3065–3078, 2005 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases