

Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma
- PMID: 22072639
- PMCID: PMC3319004
- DOI: 10.1126/scitranslmed.3002950
Differential inhibitor sensitivity of anaplastic lymphoma kinase variants found in neuroblastoma
Abstract
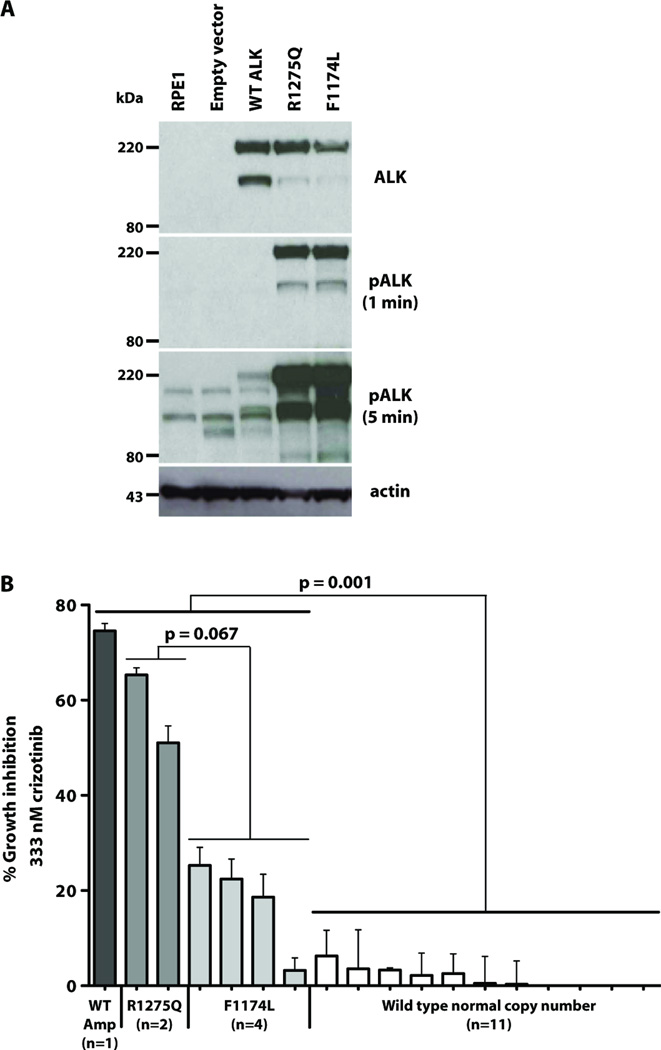
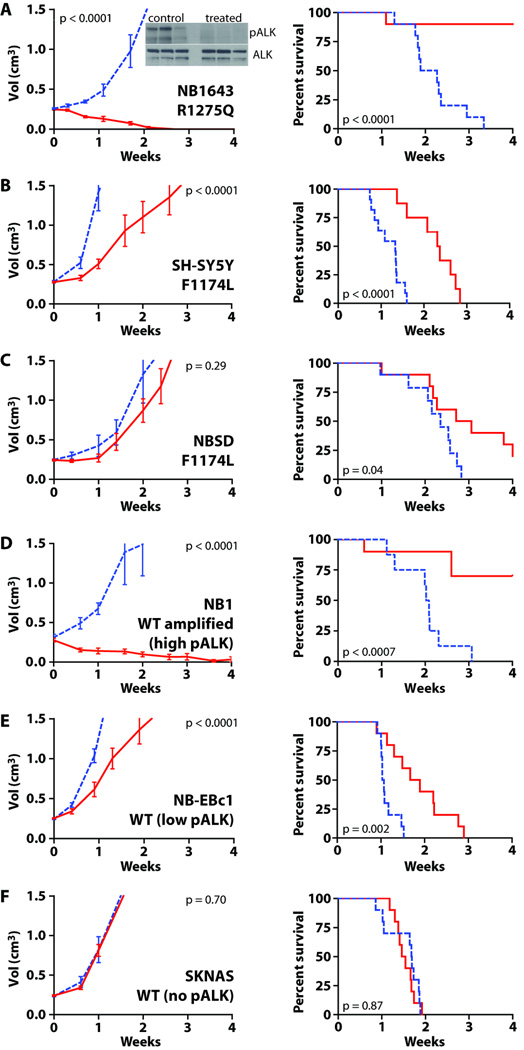
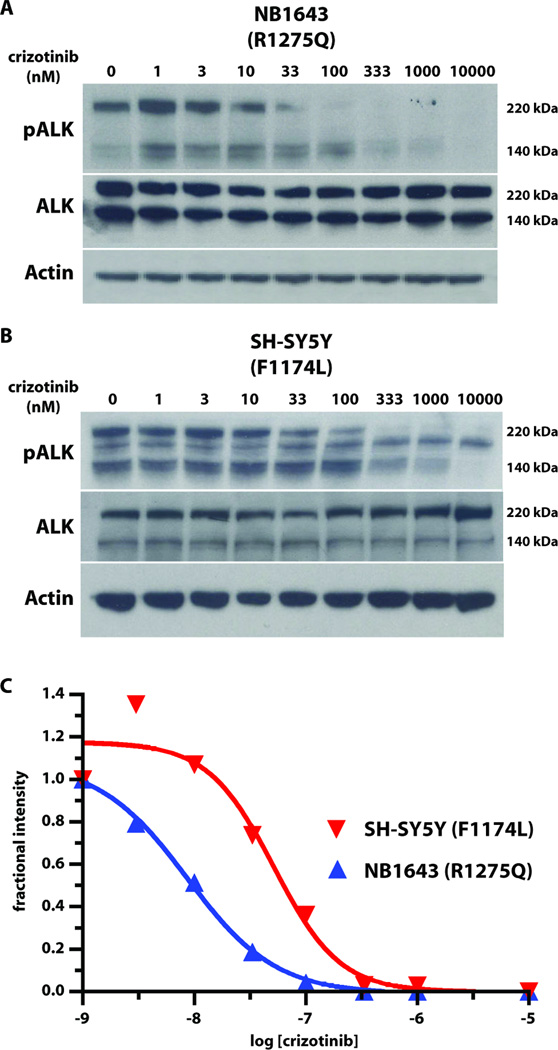
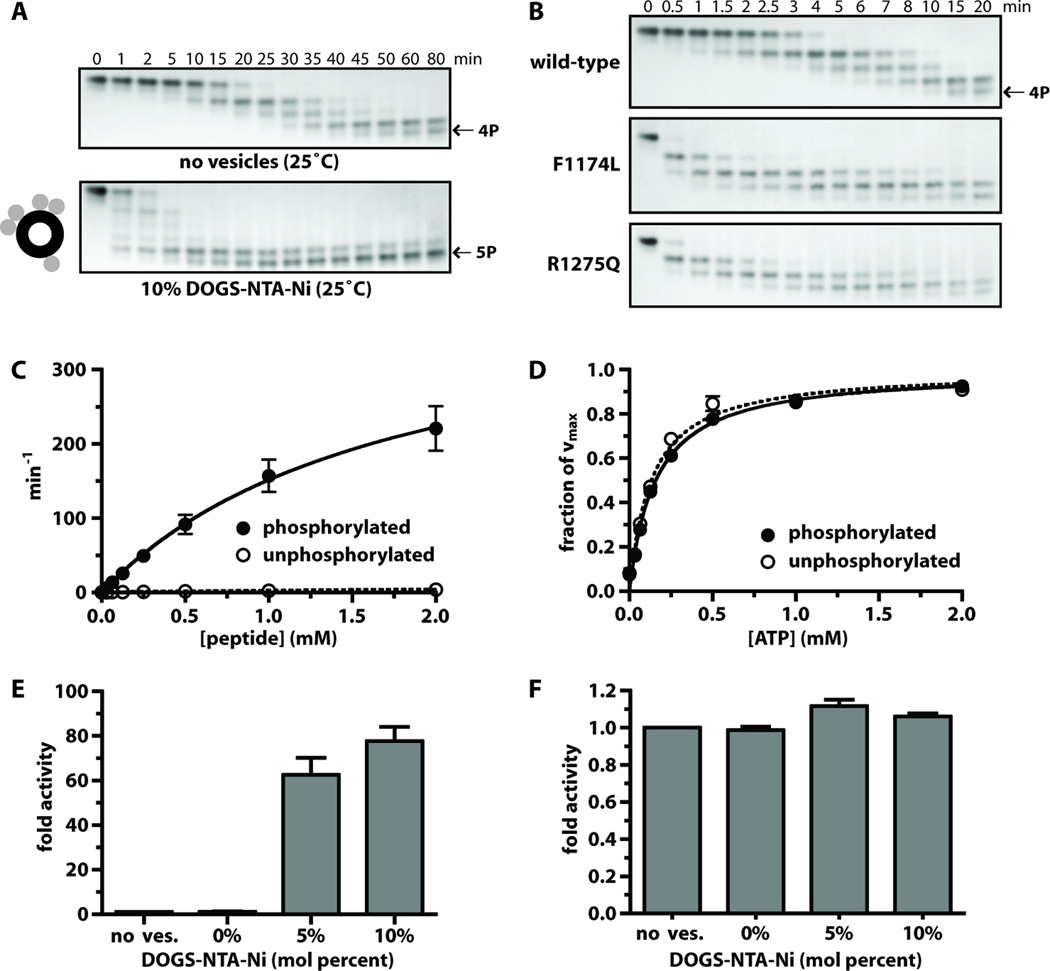
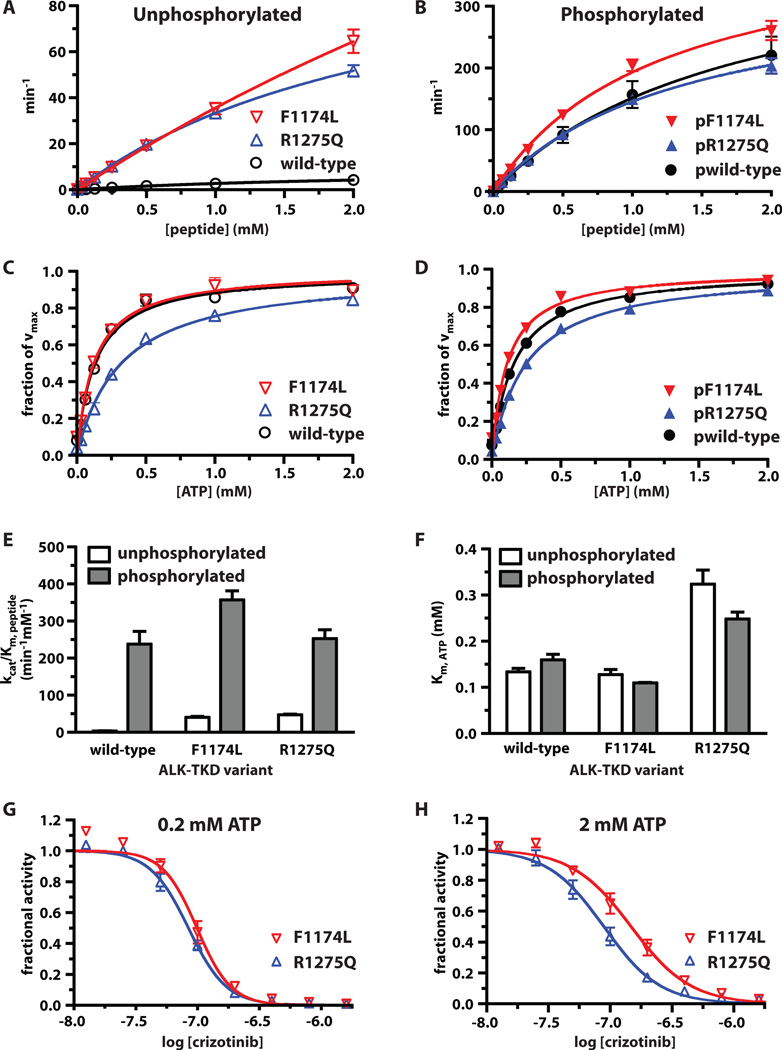
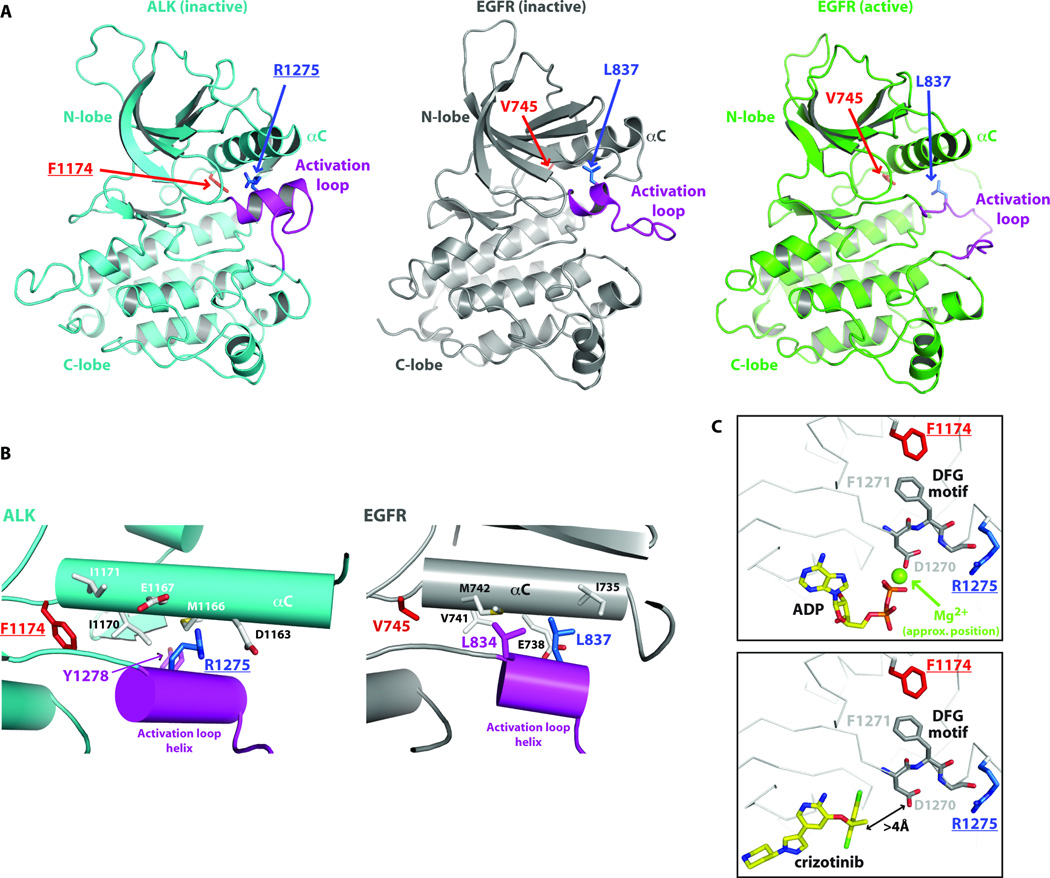
Activating mutations in the anaplastic lymphoma kinase (ALK) gene were recently discovered in neuroblastoma, a cancer of the developing autonomic nervous system that is the most commonly diagnosed malignancy in the first year of life. The most frequent ALK mutations in neuroblastoma cause amino acid substitutions (F1174L and R1275Q) in the intracellular tyrosine kinase domain of the intact ALK receptor. Identification of ALK as an oncogenic driver in neuroblastoma suggests that crizotinib (PF-02341066), a dual-specific inhibitor of the ALK and Met tyrosine kinases, will be useful in treating this malignancy. Here, we assessed the ability of crizotinib to inhibit proliferation of neuroblastoma cell lines and xenografts expressing mutated or wild-type ALK. Crizotinib inhibited proliferation of cell lines expressing either R1275Q-mutated ALK or amplified wild-type ALK. In contrast, cell lines harboring F1174L-mutated ALK were relatively resistant to crizotinib. Biochemical analyses revealed that this reduced susceptibility of F1174L-mutated ALK to crizotinib inhibition resulted from an increased adenosine triphosphate-binding affinity (as also seen in acquired resistance to epidermal growth factor receptor inhibitors). Thus, this effect should be surmountable with higher doses of crizotinib and/or with higher-affinity inhibitors.
Figures
References
-
- D'Angio GJ, Evans AE, Koop CE. Special pattern of widespread neuroblastoma with a favourable prognosis. Lancet. 1971;1:1046–1049. - PubMed
-
- Iwahara T, Fujimoto J, Wen D, Cupples R, Bucay N, Arakawa T, Mori S, Ratzkin B, Yamamoto T. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14:439–449. - PubMed
-
- Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, Look AT. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263:1281–1284. - PubMed
-
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
