

Fatal prion disease in a mouse model of genetic E200K Creutzfeldt-Jakob disease
- PMID: 22072968
- PMCID: PMC3207931
- DOI: 10.1371/journal.ppat.1002350
Fatal prion disease in a mouse model of genetic E200K Creutzfeldt-Jakob disease
Erratum in
-
Correction: Fatal Prion Disease in a Mouse Model of Genetic E200K Creutzfeldt-Jakob Disease.PLoS Pathog. 2017 May 3;13(5):e1006294. doi: 10.1371/journal.ppat.1006294. eCollection 2017 May. PLoS Pathog. 2017. PMID: 28467504 Free PMC article.
Retraction in
-
Retraction: Fatal Prion Disease in a Mouse Model of Genetic E200K Creutzfeldt-Jakob Disease.PLoS Pathog. 2024 Dec 11;20(12):e1012774. doi: 10.1371/journal.ppat.1012774. eCollection 2024 Dec. PLoS Pathog. 2024. PMID: 39661602 Free PMC article. No abstract available.
Abstract
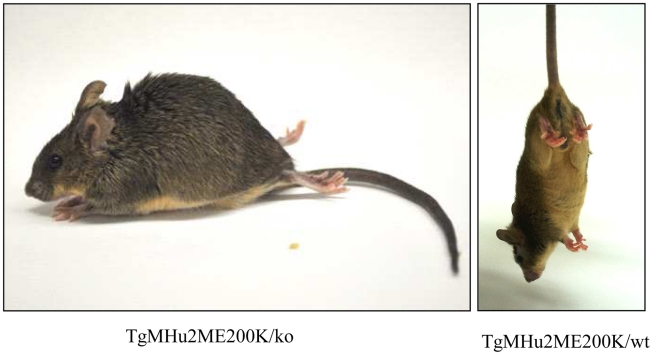
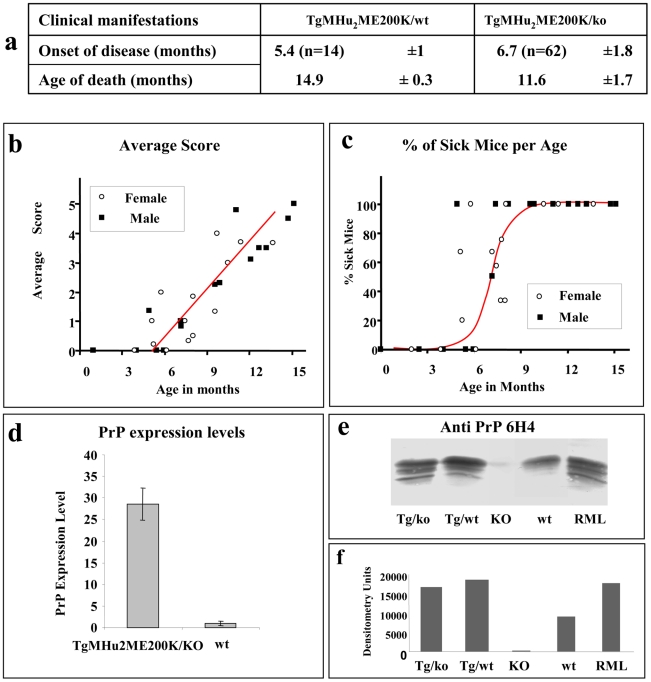
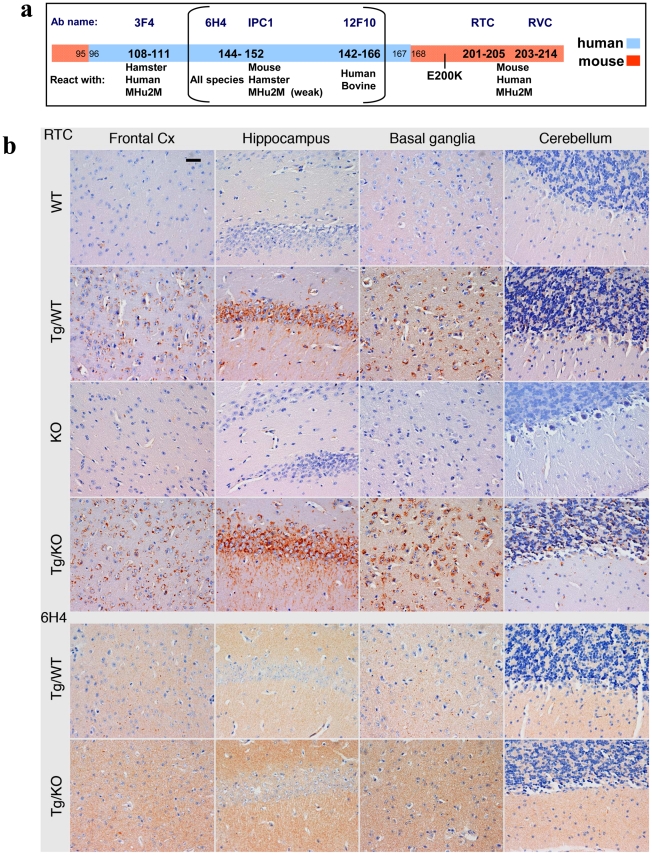
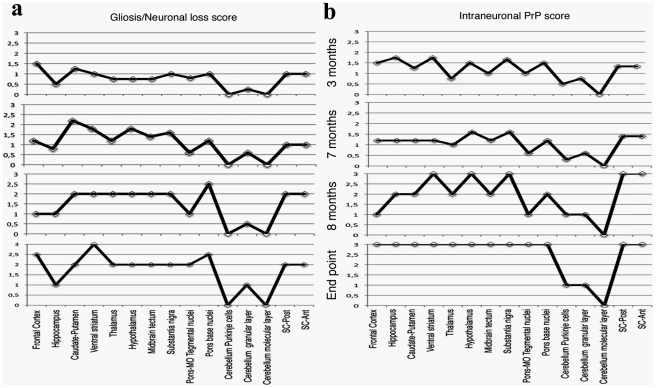
Genetic prion diseases are late onset fatal neurodegenerative disorders linked to pathogenic mutations in the prion protein-encoding gene, PRNP. The most prevalent of these is the substitution of Glutamate for Lysine at codon 200 (E200K), causing genetic Creutzfeldt-Jakob disease (gCJD) in several clusters, including Jews of Libyan origin. Investigating the pathogenesis of genetic CJD, as well as developing prophylactic treatments for young asymptomatic carriers of this and other PrP mutations, may well depend upon the availability of appropriate animal models in which long term treatments can be evaluated for efficacy and toxicity. Here we present the first effective mouse model for E200KCJD, which expresses chimeric mouse/human (TgMHu2M) E199KPrP on both a null and a wt PrP background, as is the case for heterozygous patients and carriers. Mice from both lines suffered from distinct neurological symptoms as early as 5-6 month of age and deteriorated to death several months thereafter. Histopathological examination of the brain and spinal cord revealed early gliosis and age-related intraneuronal deposition of disease-associated PrP similarly to human E200K gCJD. Concomitantly we detected aggregated, proteinase K resistant, truncated and oxidized PrP forms on immunoblots. Inoculation of brain extracts from TgMHu2ME199K mice readily induced, the first time for any mutant prion transgenic model, a distinct fatal prion disease in wt mice. We believe that these mice may serve as an ideal platform for the investigation of the pathogenesis of genetic prion disease and thus for the monitoring of anti-prion treatments.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
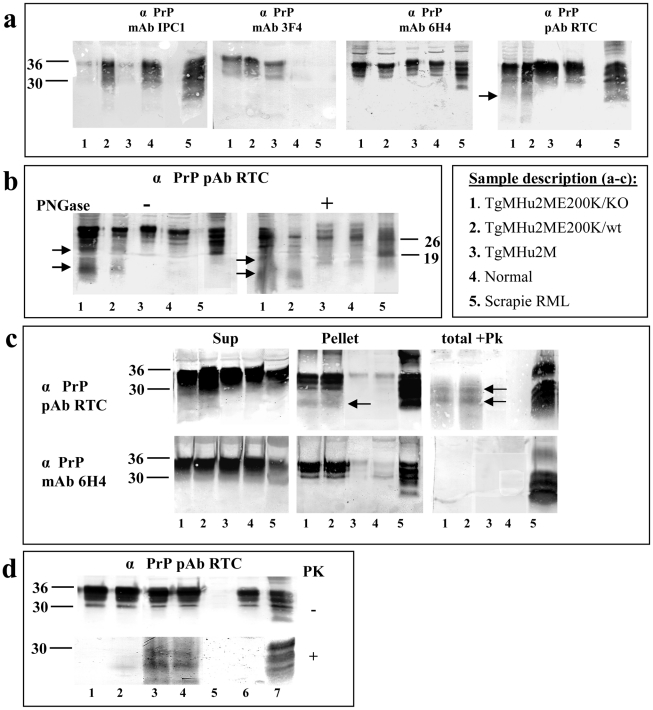
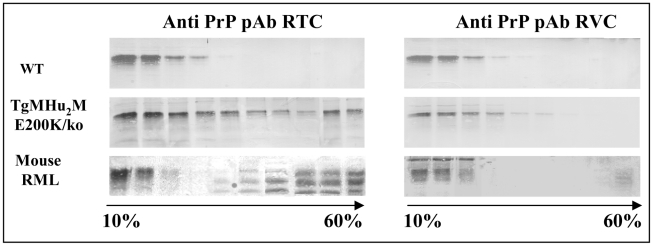
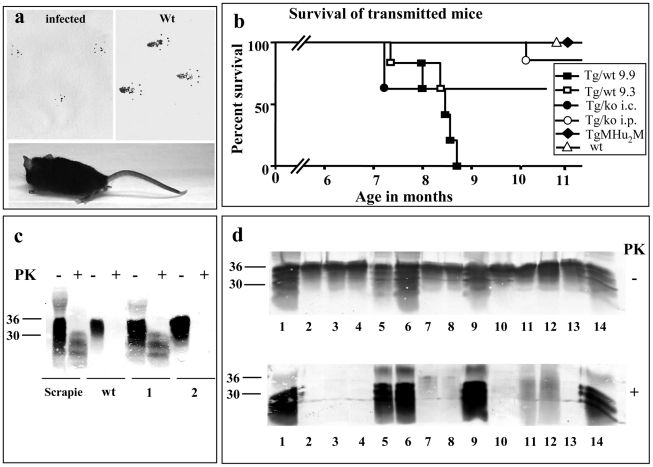
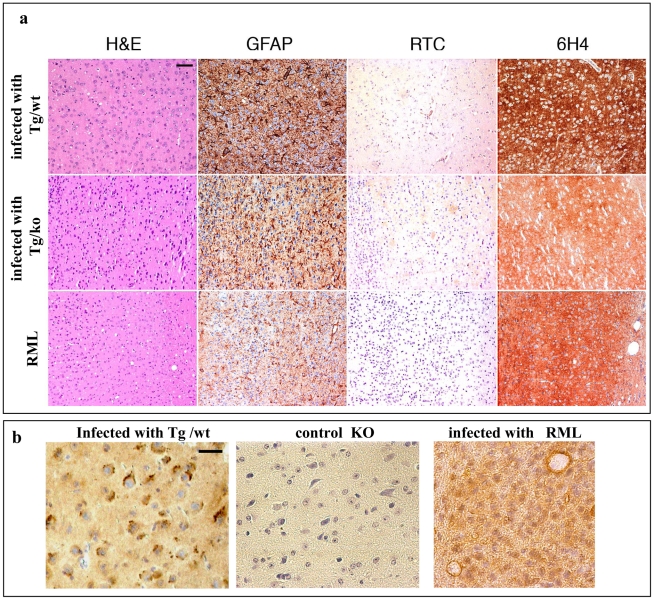
References
-
- Kovacs GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, et al. Genetic prion disease: the EUROCJD experience. Hum Genet. 2005;118:166–174. - PubMed
-
- Hsiao K, Prusiner SB. Inherited human prion diseases. Neurology. 1990;40:1820–1827. - PubMed
-
- Hsiao K, Meiner Z, Kahana E, Cass C, Kahana I, et al. Mutation of the prion protein in Libyan Jews with Creutzfeldt-Jakob disease. N Engl J Med. 1991;324:1091–1097. - PubMed
-
- Korczyn AD, Chapman J, Goldfarb LG, Brown P, Gajdusek DC. A mutation in the prion protein gene in Creutzfeldt-Jakob disease in Jewish patients of Libyan, Greek, and Tunisian origin. Ann N Y Acad Sci. 1991;640:171–176. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
