

Cellular and molecular mechanisms in environmental and occupational inhalation toxicology
- PMID: 22073044
- PMCID: PMC3199796
Cellular and molecular mechanisms in environmental and occupational inhalation toxicology
Abstract
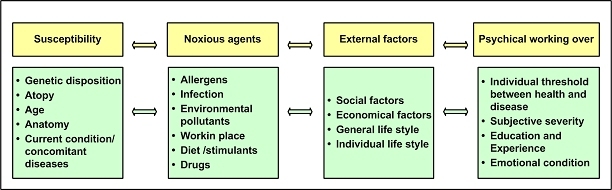
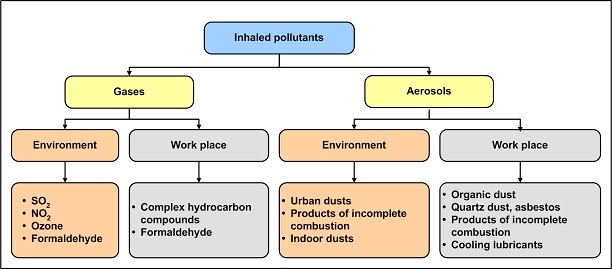
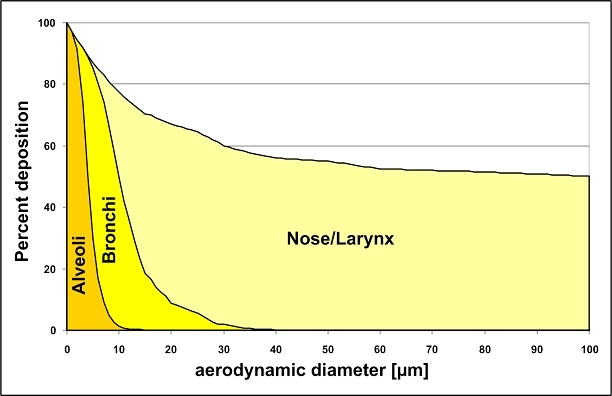
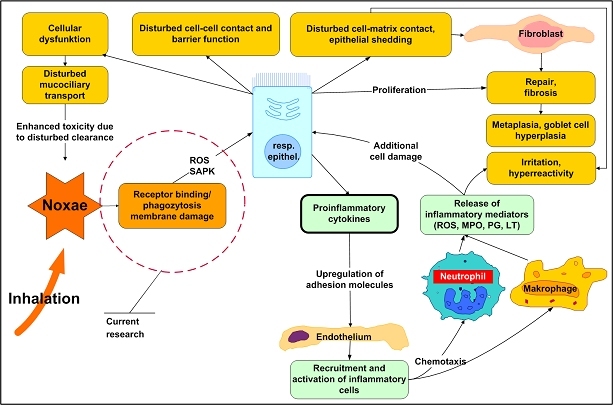
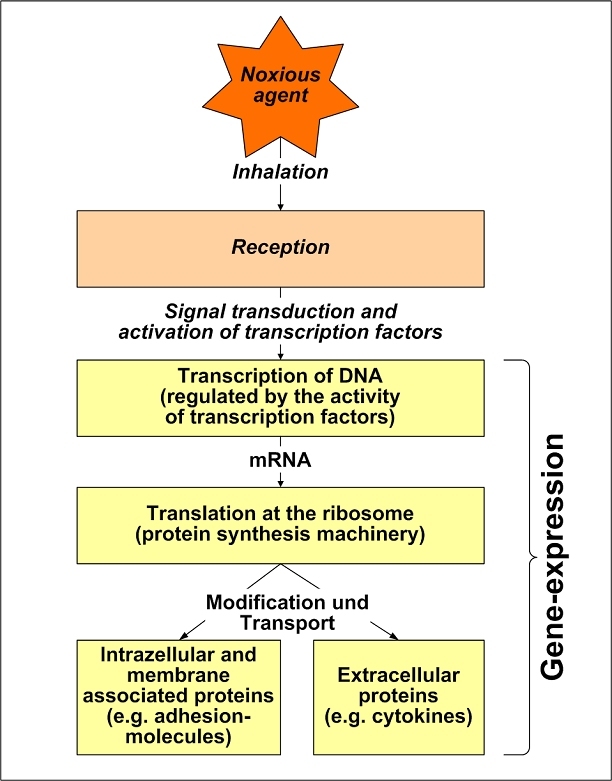
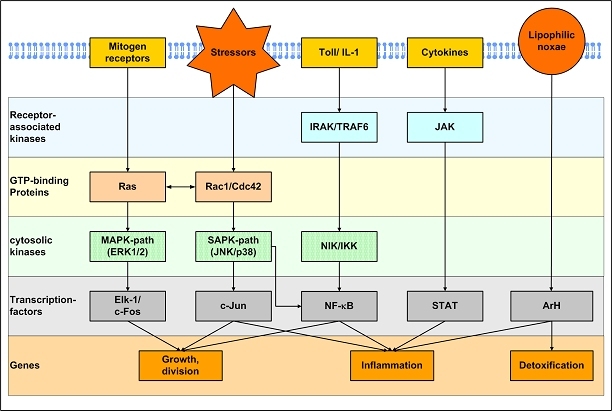
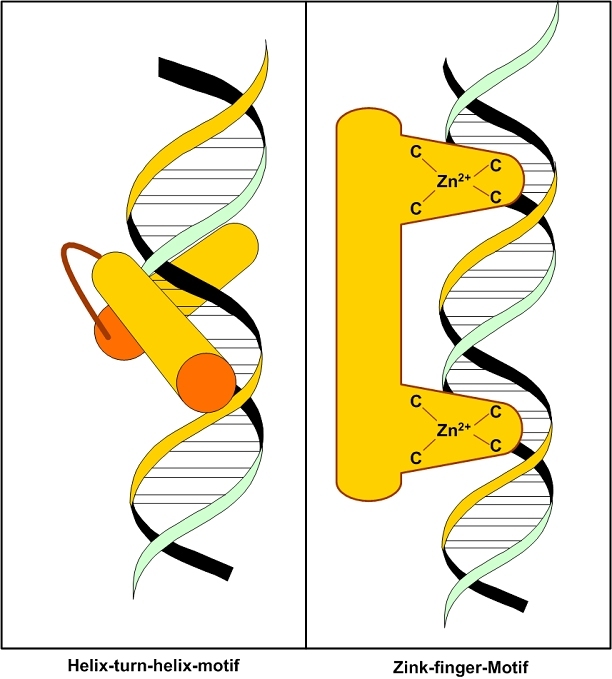
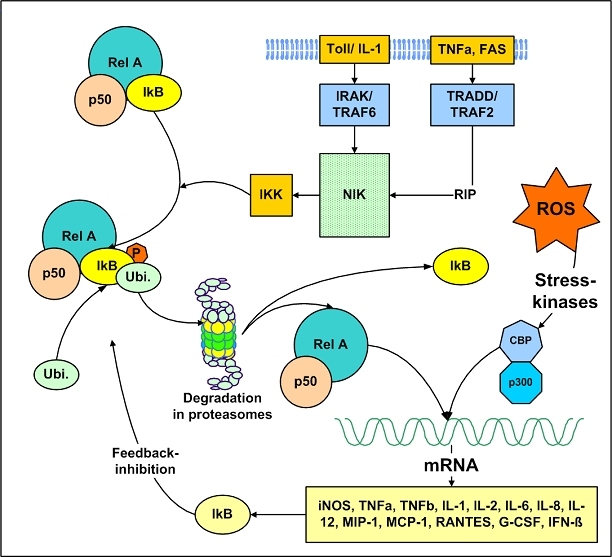
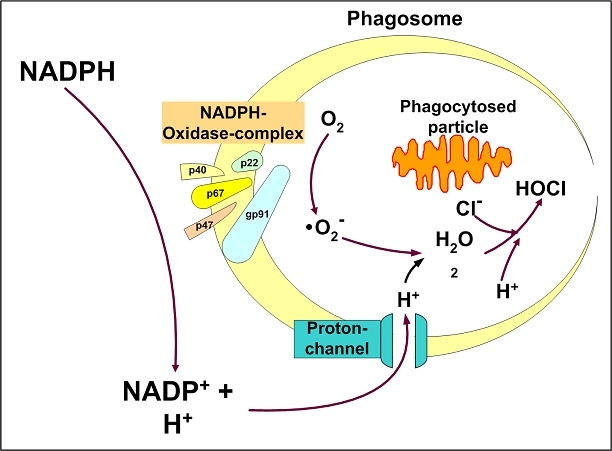
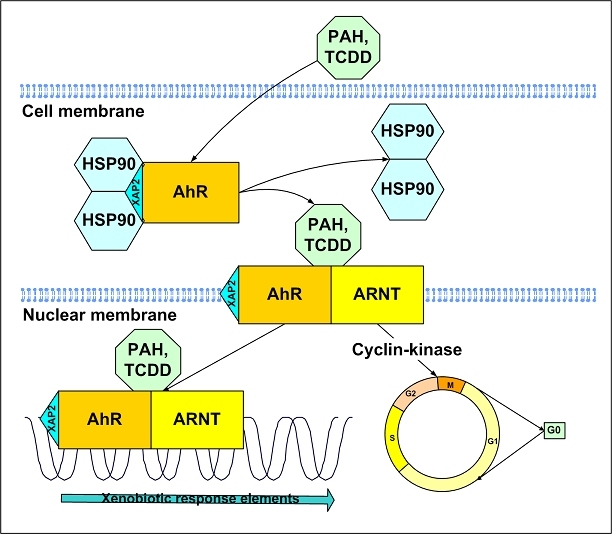
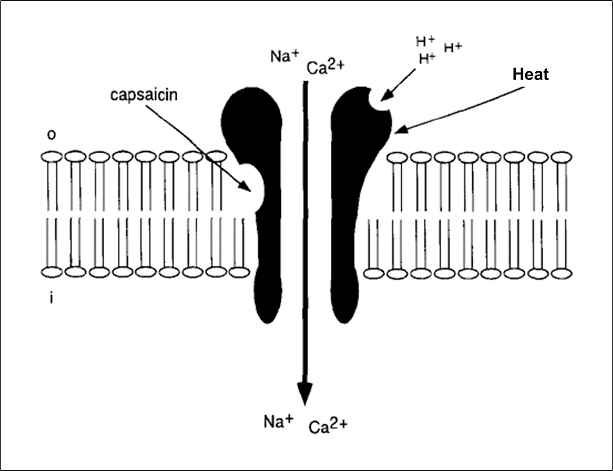
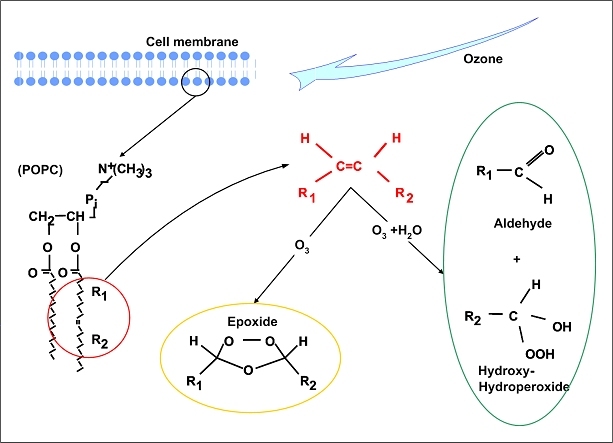
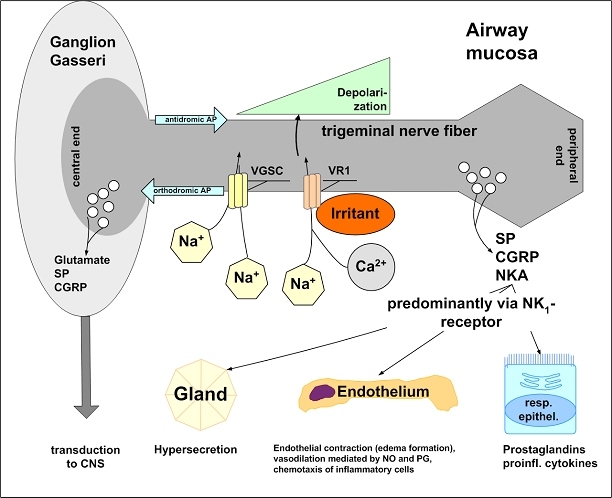
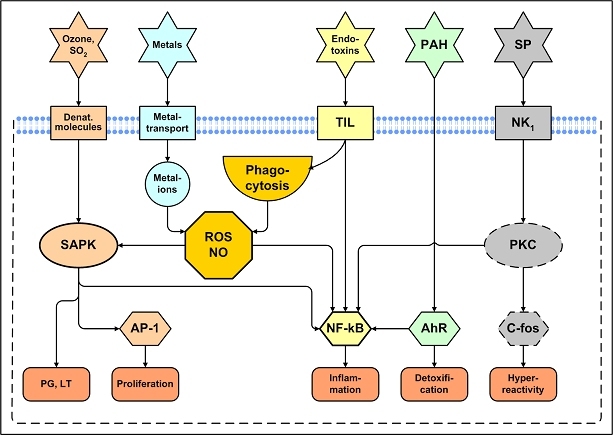
The central issue of this review are inflammatory changes that take place in the mucous membranes of the respiratory tract as a result of inhaled pollutants. Of particular relevance are dusts, SO(2), ozone, aldehydes und volatile organic compounds. Bioorganic pollutants, especially fragments of bacteria and fungi, occur predominantly in indoor dusts. They activate the toll-like/IL-1 receptor and lead to the activation of the transcription factor NF-κB for the release of numerous proinflammatory cytokines. Metals are predominant in ambient air dust particles. They induce the release of reactive oxygen species that cause damage to lipids, proteins and the DNA of the cell. As well as NF-κB, transcription factors that foster proliferation are activated via stress activated protein kinases. Organic compounds such as polycyclic aromatic hydrocarbons and nitroso-compounds of incomplete combustion processes activate additional via the cytosolic arylhydrocarbon receptor for detoxification enzymes. Sulphur dioxide leads to acid stress, and ozone to oxidative stress of the cell. This is accompanied by the release of proinflammatory cytokines via stress activated protein kinases. Aldehydes and volatile organic compounds activate the vanilloid receptor of trigeminal nerve fibres and induce a hyperreactivity of the mucous membrane via the release of nerve growth factors. The mechanisms described work synergistically and lead to a chronic inflammatory reaction of the mucous membranes of the upper respiratory tract that is regularly demonstrable in inhabitants of western industrial nations. It is unclear whether we are dealing here with a physiological inflammation or with an at least partially avoidable result of chronic pollutant exposure.
Keywords: cell membrane receptors; endotoxins; metals; nitrogen monoxide; polycyclic aromatic hydrocarbons; reactive oxygen species; respiratory mucous membrane; signal transduction; toxicology; transcription factors.
Figures




References
-
- von-Mutius E, Sherrill DL, Fritzsch C, Martinez FD, Lebowitz MD. Air pollution and upper respiratory symptoms in children from East Germany. Eur Respir J. 1995;8:723–728. - PubMed
-
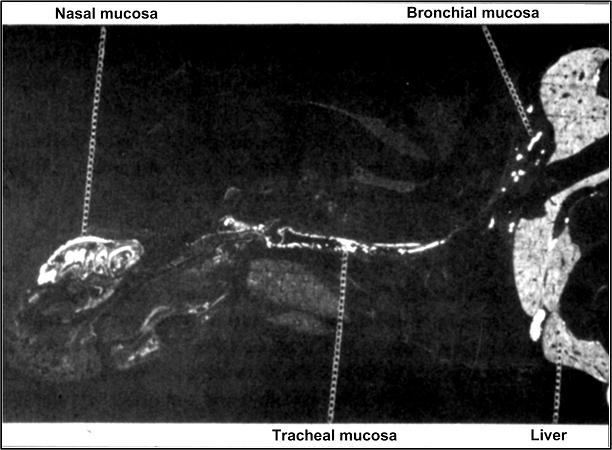
- Calderon-Garciduenas L, Rodriguez-Alcaraz A, Villarreal-Calderon A, Lyght O, Janszen D, Morgan KT. Nasal epithelium as a sentinel for airborne environmental pollution. Toxicol Sci. 1998;46:352–364. - PubMed
-
- Keles N, Ilicali OC, Deger K. Impact of air pollution on prevalence of rhinitis in Istanbul. Arch Environ Health. 1999;54:48–51. - PubMed
-
- Keles N, Ilicali C. The impact of outdoor pollution on upper respiratory diseases. Rhinology. 1998;36:24–27. - PubMed
-
- Abbey DE, Hwang BL, Burchette RJ, Vancuren T, Mills PK. Estimated long-term ambient concentrations of PM10 and development of respiratory symptoms in a nonsmoking population. Arch Environ Health. 1995;50:139–152. - PubMed
LinkOut - more resources
Full Text Sources