

NNAlign: a web-based prediction method allowing non-expert end-user discovery of sequence motifs in quantitative peptide data
- PMID: 22073191
- PMCID: PMC3206854
- DOI: 10.1371/journal.pone.0026781
NNAlign: a web-based prediction method allowing non-expert end-user discovery of sequence motifs in quantitative peptide data
Abstract
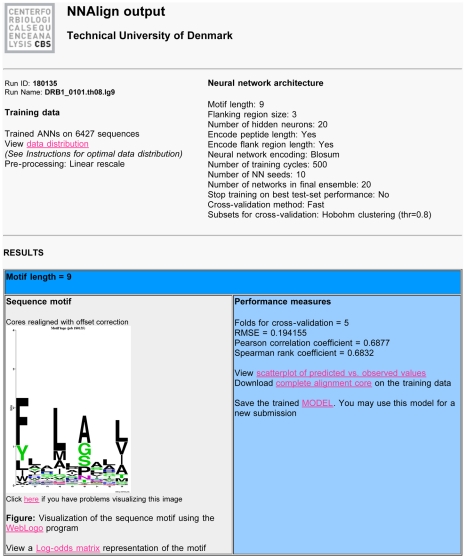
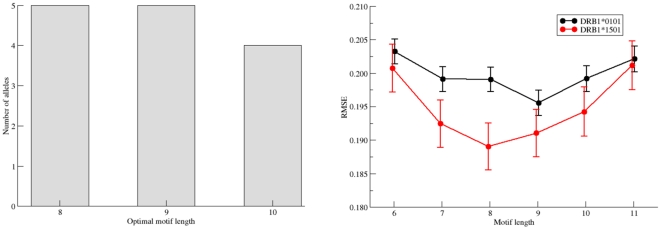
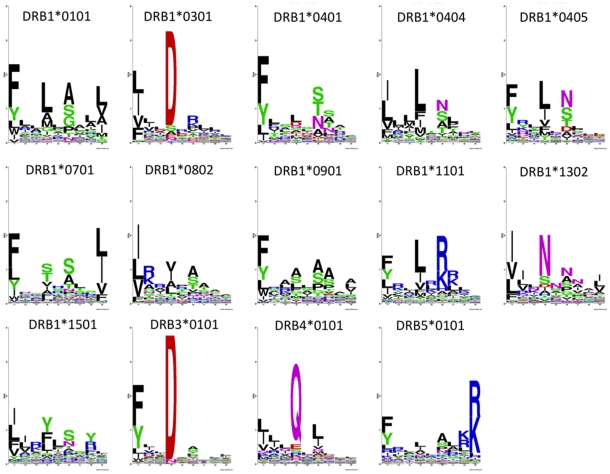
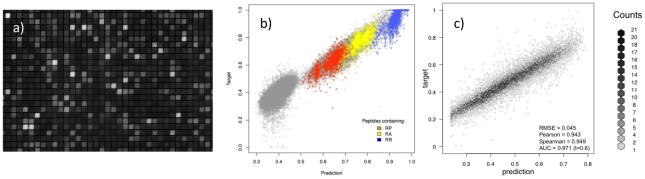
Recent advances in high-throughput technologies have made it possible to generate both gene and protein sequence data at an unprecedented rate and scale thereby enabling entirely new "omics"-based approaches towards the analysis of complex biological processes. However, the amount and complexity of data that even a single experiment can produce seriously challenges researchers with limited bioinformatics expertise, who need to handle, analyze and interpret the data before it can be understood in a biological context. Thus, there is an unmet need for tools allowing non-bioinformatics users to interpret large data sets. We have recently developed a method, NNAlign, which is generally applicable to any biological problem where quantitative peptide data is available. This method efficiently identifies underlying sequence patterns by simultaneously aligning peptide sequences and identifying motifs associated with quantitative readouts. Here, we provide a web-based implementation of NNAlign allowing non-expert end-users to submit their data (optionally adjusting method parameters), and in return receive a trained method (including a visual representation of the identified motif) that subsequently can be used as prediction method and applied to unknown proteins/peptides. We have successfully applied this method to several different data sets including peptide microarray-derived sets containing more than 100,000 data points. NNAlign is available online at http://www.cbs.dtu.dk/services/NNAlign.
Conflict of interest statement
Figures

References
-
- James W. Nucleic acid and polypeptide aptamers: a powerful approach to ligand discovery. Curr Opin Pharmacol. 2001;1:540–546. - PubMed
-
- Hoppe-Seyler F, Crnkovic-Mertens I, Tomai E, Butz K. Peptide aptamers: specific inhibitors of protein function. Curr Mol Med. 2004;4:529–538. - PubMed
-
- Schutkowski M, Reineke U, Reimer U. Peptide arrays for kinase profiling. Chembiochem. 2005;6:513–521. - PubMed
-
- Han X, Yamanouchi G, Mori T, Kang JH, Niidome T, et al. Monitoring protein kinase activity in cell lysates using a high-density peptide microarray. J Biomol Screen. 2009;14:256–262. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
