

The ATM protein kinase and cellular redox signaling: beyond the DNA damage response
- PMID: 22079189
- PMCID: PMC3259275
- DOI: 10.1016/j.tibs.2011.10.002
The ATM protein kinase and cellular redox signaling: beyond the DNA damage response
Abstract
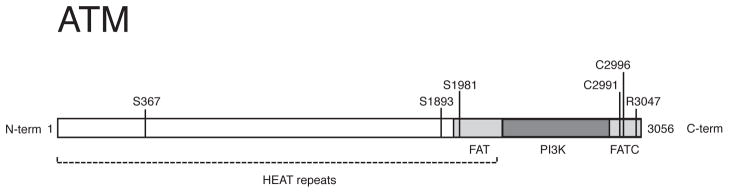
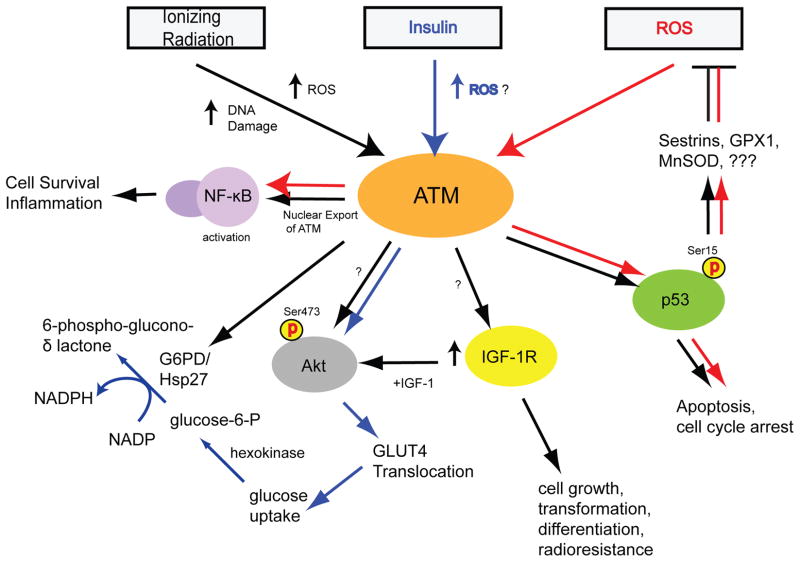
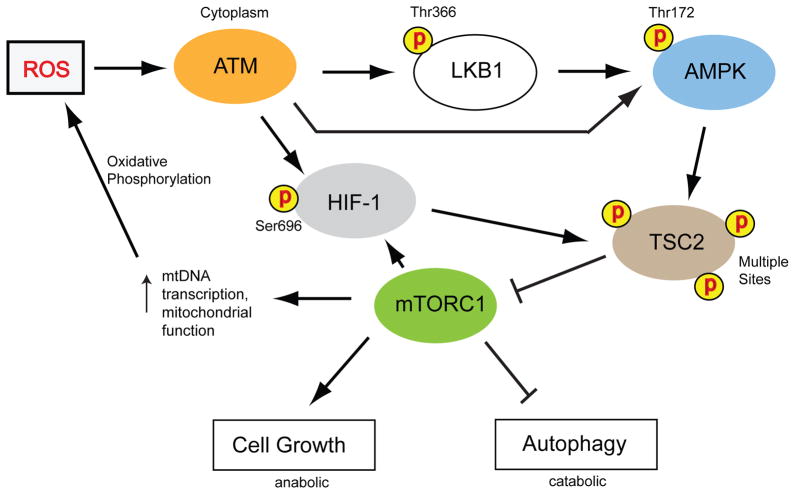
The ataxia-telangiectasia mutated (ATM) protein kinase is best known for its role in the DNA damage response, but recent findings suggest that it also functions as a redox sensor that controls the levels of reactive oxygen species in human cells. Here, we review evidence supporting the conclusion that ATM can be directly activated by oxidation, as well as various observations from ATM-deficient patients and mouse models that point to the importance of ATM in oxidative stress responses. We also discuss the roles of this kinase in regulating mitochondrial function and metabolic control through its action on tumor suppressor p53, AMP-activated protein kinase (AMPK), mammalian target of rapamycin (mTOR) and hypoxia-inducible factor 1 (HIF1), and how the regulation of these enzymes may be affected in ATM-deficient patients and in cancer cells.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
References
-
- Lavin MF, et al. Current and potential therapeutic strategies for the treatment of ataxia-telangiectasia. Br Med Bull. 2007;81–82:129–147. - PubMed
-
- Ristow M. Neurodegenerative disorders associated with diabetes mellitus. J Mol Med (Berl) 2004;82:510–529. - PubMed
-
- Bredemeyer AL, et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 2006;442:466–470. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
