

Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity
- PMID: 22080866
- PMCID: PMC3226000
- DOI: 10.1172/JCI58577
Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity
Abstract
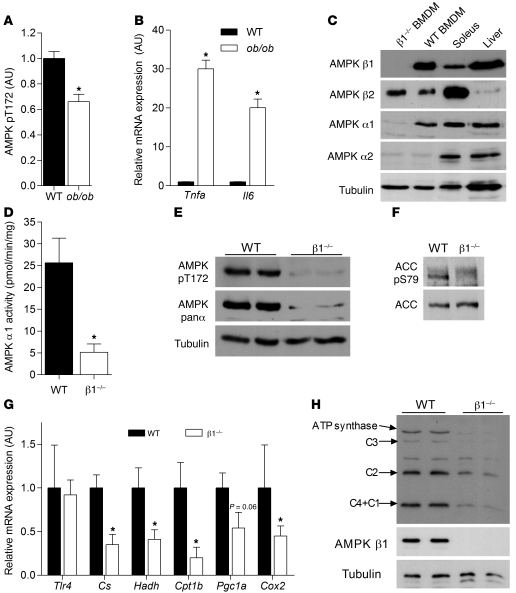
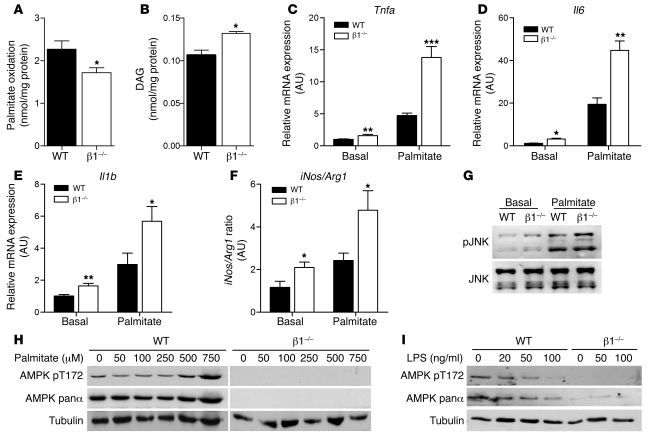
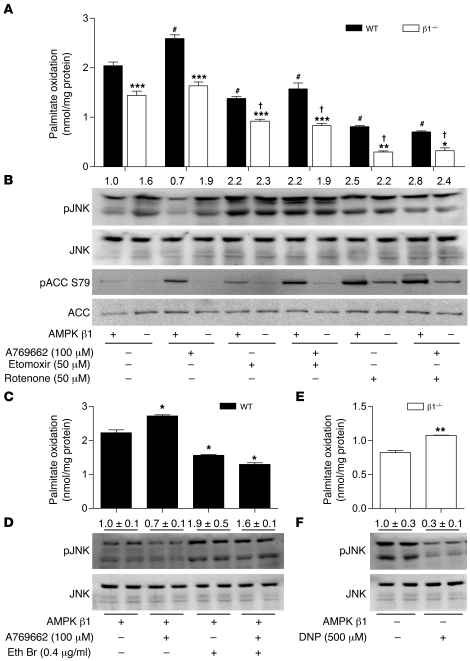
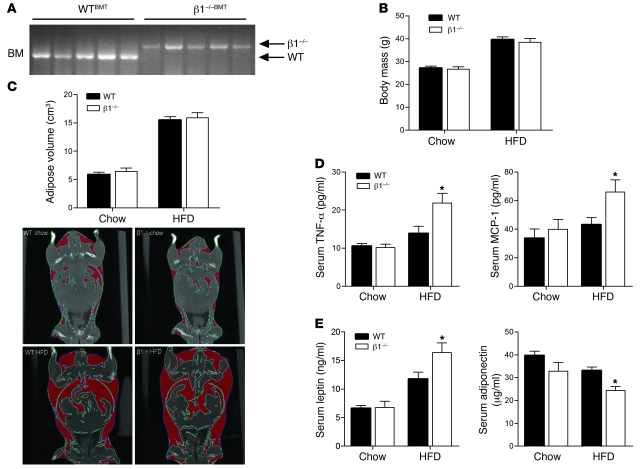
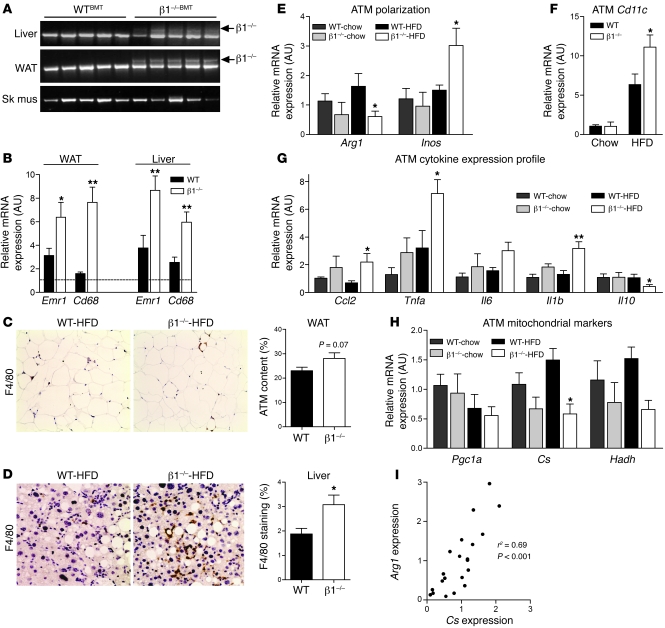
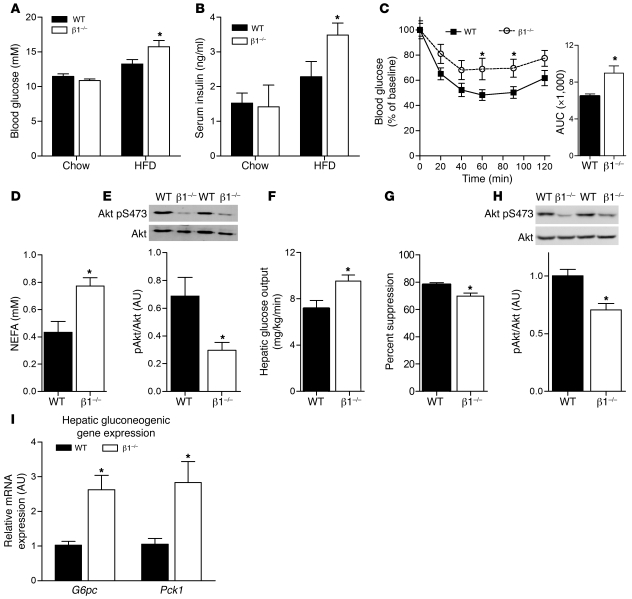
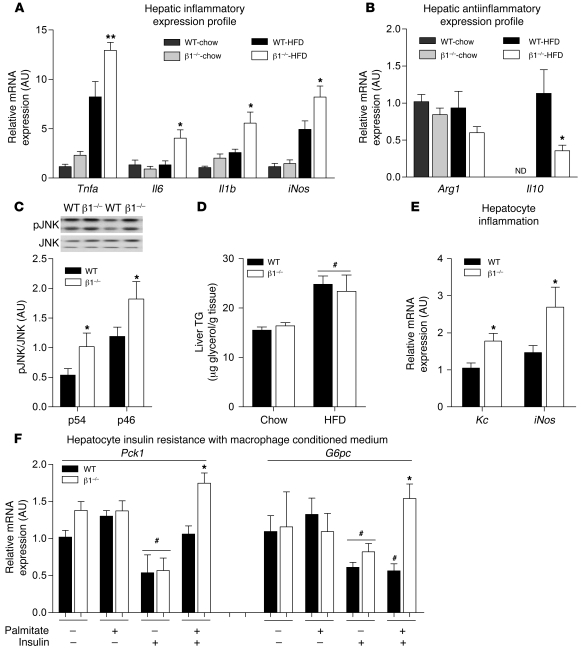
Individuals who are obese are frequently insulin resistant, putting them at increased risk of developing type 2 diabetes and its associated adverse health conditions. The accumulation in adipose tissue of macrophages in an inflammatory state is a hallmark of obesity-induced insulin resistance. Here, we reveal a role for AMPK β1 in protecting macrophages from inflammation under high lipid exposure. Genetic deletion of the AMPK β1 subunit in mice (referred to herein as β1(-/-) mice) reduced macrophage AMPK activity, acetyl-CoA carboxylase phosphorylation, and mitochondrial content, resulting in reduced rates of fatty acid oxidation. β1(-/-) macrophages displayed increased levels of diacylglycerol and markers of inflammation, effects that were reproduced in WT macrophages by inhibiting fatty acid oxidation and, conversely, prevented by pharmacological activation of AMPK β1-containing complexes. The effect of AMPK β1 loss in macrophages was tested in vivo by transplantation of bone marrow from WT or β1(-/-) mice into WT recipients. When challenged with a high-fat diet, mice that received β1(-/-) bone marrow displayed enhanced adipose tissue macrophage inflammation and liver insulin resistance compared with animals that received WT bone marrow. Thus, activation of AMPK β1 and increasing fatty acid oxidation in macrophages may represent a new therapeutic approach for the treatment of insulin resistance.
Figures
Comment in
-
AMPK regulates macrophage polarization in adipose tissue inflammation and NASH.J Hepatol. 2013 Mar;58(3):619-21. doi: 10.1016/j.jhep.2012.09.031. Epub 2012 Oct 6. J Hepatol. 2013. PMID: 23046670 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
