

Antioxidant therapies in traumatic brain and spinal cord injury
- PMID: 22080976
- PMCID: PMC4134010
- DOI: 10.1016/j.bbadis.2011.10.017
Antioxidant therapies in traumatic brain and spinal cord injury
Abstract
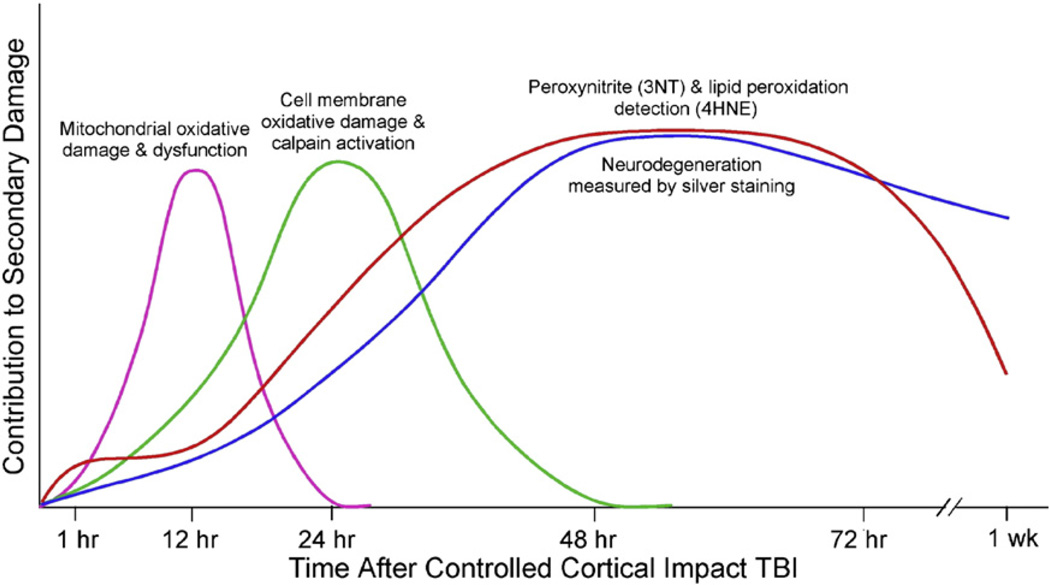
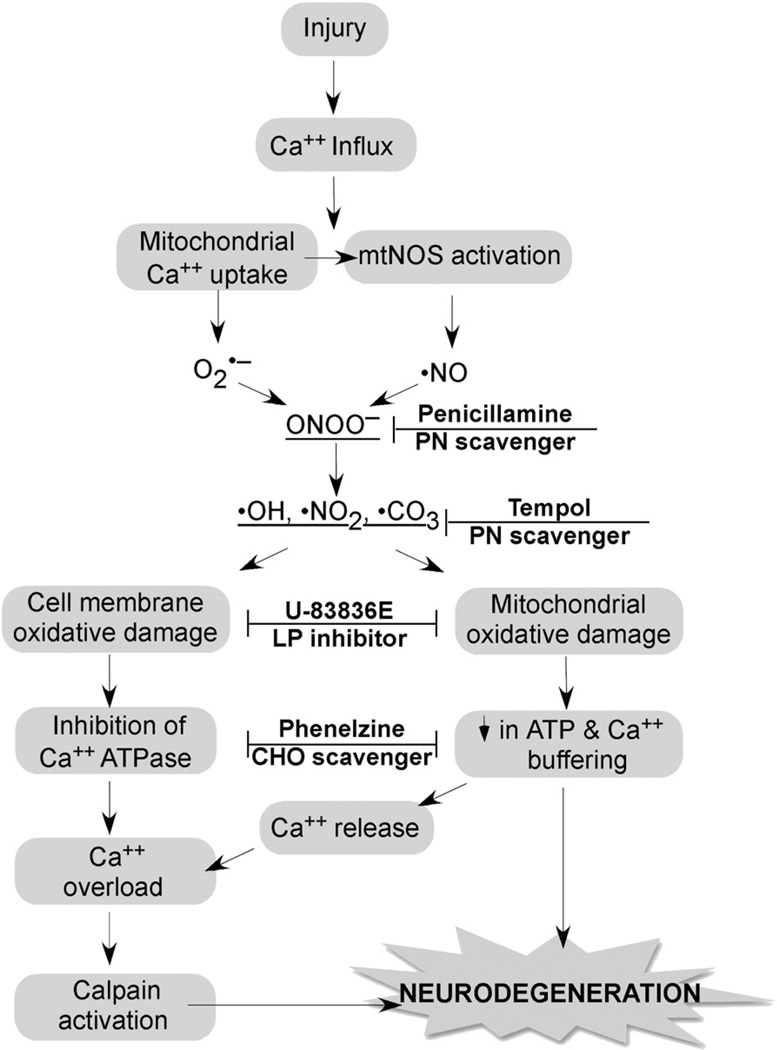
Free radical formation and oxidative damage have been extensively investigated and validated as important contributors to the pathophysiology of acute central nervous system injury. The generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) is an early event following injury occurring within minutes of mechanical impact. A key component in this event is peroxynitrite-induced lipid peroxidation. As discussed in this review, peroxynitrite formation and lipid peroxidation irreversibly damages neuronal membrane lipids and protein function, which results in subsequent disruptions in ion homeostasis, glutamate-mediated excitotoxicity, mitochondrial respiratory failure and microvascular damage. Antioxidant approaches include the inhibition and/or scavenging of superoxide, peroxynitrite, or carbonyl compounds, the inhibition of lipid peroxidation and the targeting of the endogenous antioxidant defense system. This review covers the preclinical and clinical literature supporting the role of ROS and RNS and their derived oxygen free radicals in the secondary injury response following acute traumatic brain injury (TBI) and spinal cord injury (SCI) and reviews the past and current trends in the development of antioxidant therapeutic strategies. Combinatorial treatment with the suggested mechanistically complementary antioxidants will also be discussed as a promising neuroprotective approach in TBI and SCI therapeutic research. This article is part of a Special Issue entitled: Antioxidants and antioxidant treatment in disease.
Copyright © 2011 Elsevier B.V. All rights reserved.
Figures
References
-
- Beckman JS. Oxidative damage and tyrosine nitration from peroxynitrite. Chem. Res. Toxicol. 1996;9:836–844. - PubMed
-
- Radi R. Peroxynitrite reactions and diffusion in biology. Chem. Res. Toxicol. 1998;11:720–721. - PubMed
-
- Hall ED, Oostveen JA, Andrus PK, Anderson DK, Thomas CE. Immunocyto-chemical method for investigating in vivo neuronal oxygen radical-induced lipid peroxidation. J. Neurosci. Methods. 1997;76:115–122. - PubMed
-
- Gutteridge JM. Lipid peroxidation and antioxidants as biomarkers of tissue damage. Clin. Chem. 1995;41:1819–1828. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
