

Single-cell dissection of transcriptional heterogeneity in human colon tumors
- PMID: 22081019
- PMCID: PMC3237928
- DOI: 10.1038/nbt.2038
Single-cell dissection of transcriptional heterogeneity in human colon tumors
Abstract
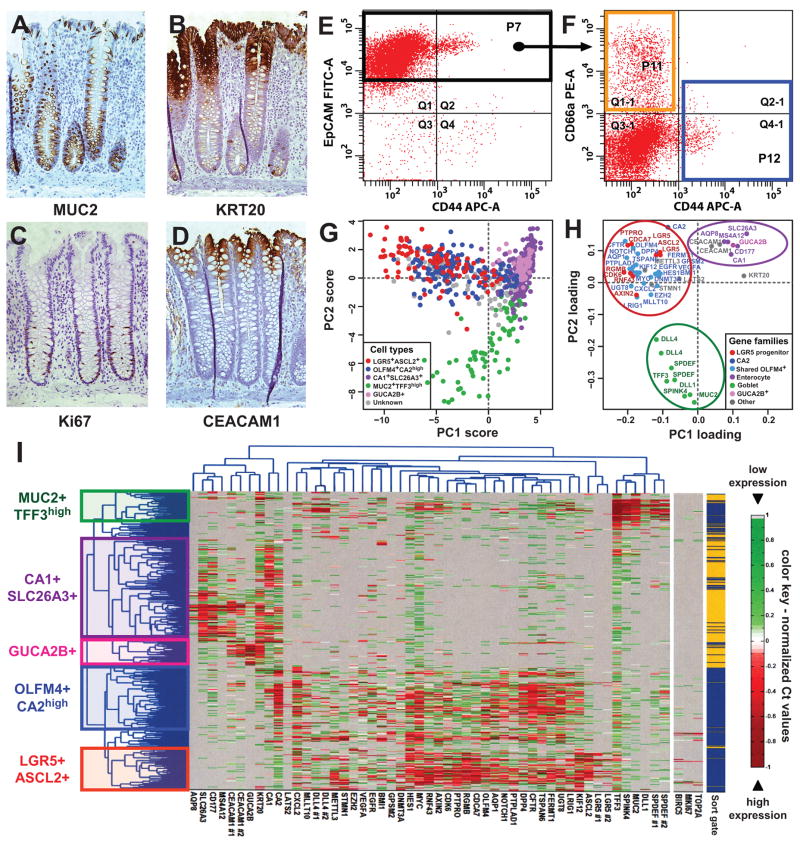
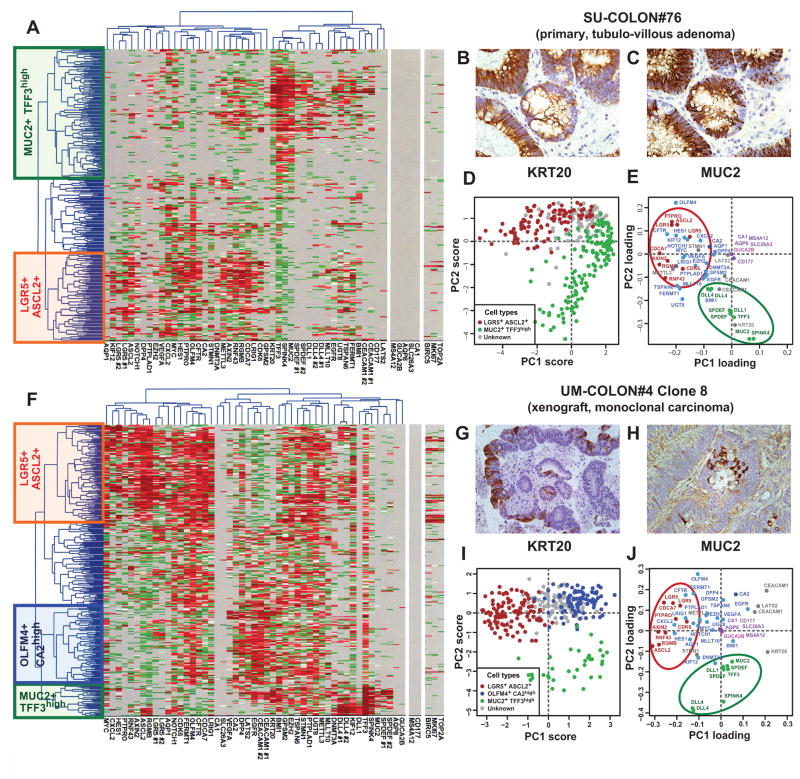
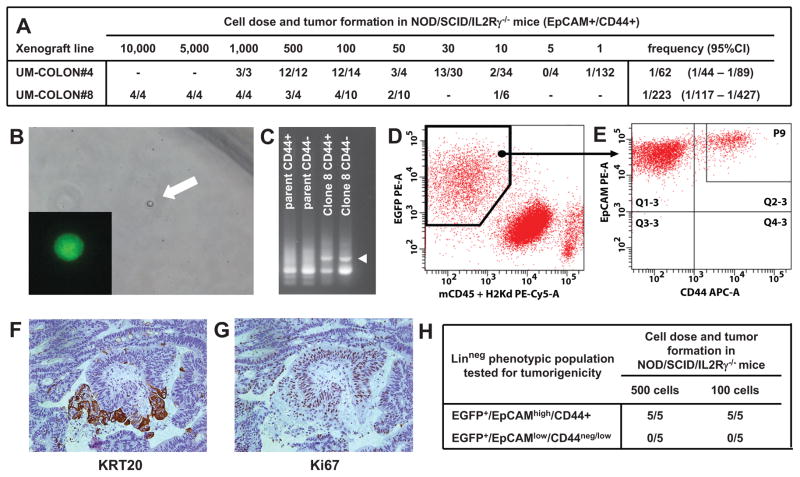
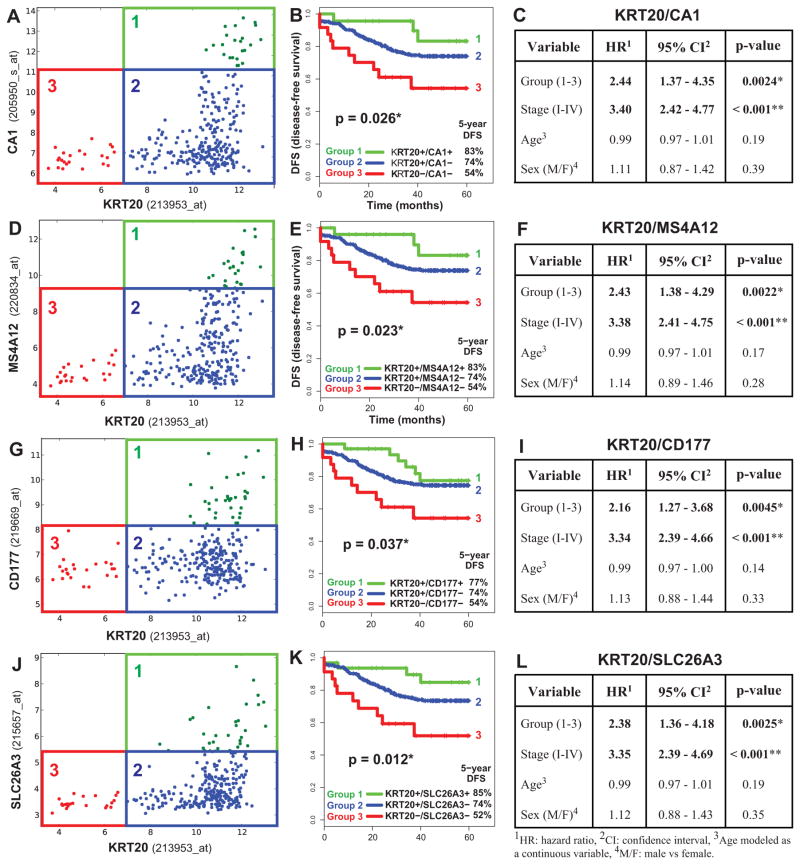
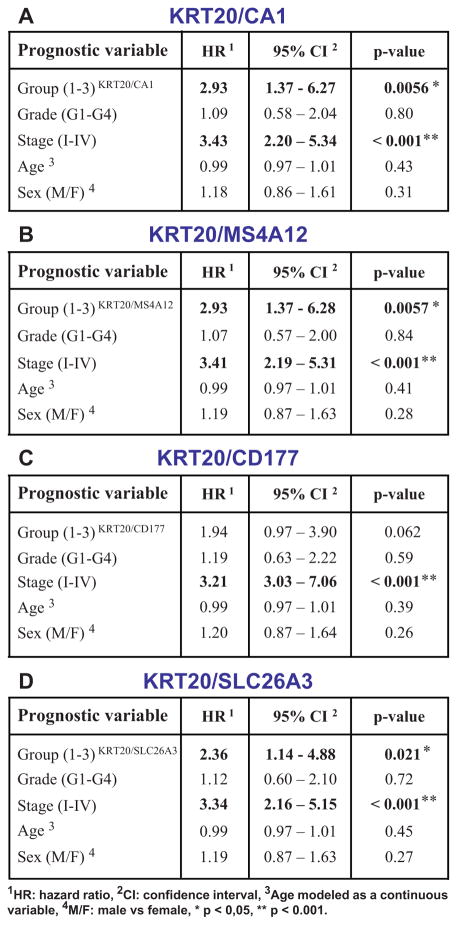
Cancer is often viewed as a caricature of normal developmental processes, but the extent to which its cellular heterogeneity truly recapitulates multilineage differentiation processes of normal tissues remains unknown. Here we implement single-cell PCR gene-expression analysis to dissect the cellular composition of primary human normal colon and colon cancer epithelia. We show that human colon cancer tissues contain distinct cell populations whose transcriptional identities mirror those of the different cellular lineages of normal colon. By creating monoclonal tumor xenografts from injection of a single (n = 1) cell, we demonstrate that the transcriptional diversity of cancer tissues is largely explained by in vivo multilineage differentiation and not only by clonal genetic heterogeneity. Finally, we show that the different gene-expression programs linked to multilineage differentiation are strongly associated with patient survival. We develop two-gene classifier systems (KRT20 versus CA1, MS4A12, CD177, SLC26A3) that predict clinical outcomes with hazard ratios superior to those of pathological grade and comparable to those of microarray-derived multigene expression signatures.
Figures
Comment in
-
Dissecting cancer heterogeneity.Nat Biotechnol. 2011 Dec 8;29(12):1095-6. doi: 10.1038/nbt.2063. Nat Biotechnol. 2011. PMID: 22158362 No abstract available.
-
Colorectal cancer: A single-cell snapshot of gene expression in healthy and cancerous colon tissue.Nat Rev Gastroenterol Hepatol. 2011 Dec 13;9(1):2. doi: 10.1038/nrgastro.2011.239. Nat Rev Gastroenterol Hepatol. 2011. PMID: 22158382 No abstract available.
-
Unraveling the transcriptional heterogeneity in human colon cancer: potential new insights regarding pathobiology and treatment.Clin Chem. 2012 Jun;58(6):966-7. doi: 10.1373/clinchem.2012.182972. Epub 2012 Apr 9. Clin Chem. 2012. PMID: 22490619 No abstract available.
References
-
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. - PubMed
-
- Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355:1253–1261. - PubMed
-
- Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–284. - PubMed
-
- Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822–829. - PubMed
-
- Campbell LL, Polyak K. Breast tumor heterogeneity: cancer stem cells or clonal evolution? Cell Cycle. 2007;6:2332–2338. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
