

Oxidation of CaMKII determines the cardiotoxic effects of aldosterone
- PMID: 22081025
- PMCID: PMC3332099
- DOI: 10.1038/nm.2506
Oxidation of CaMKII determines the cardiotoxic effects of aldosterone
Abstract
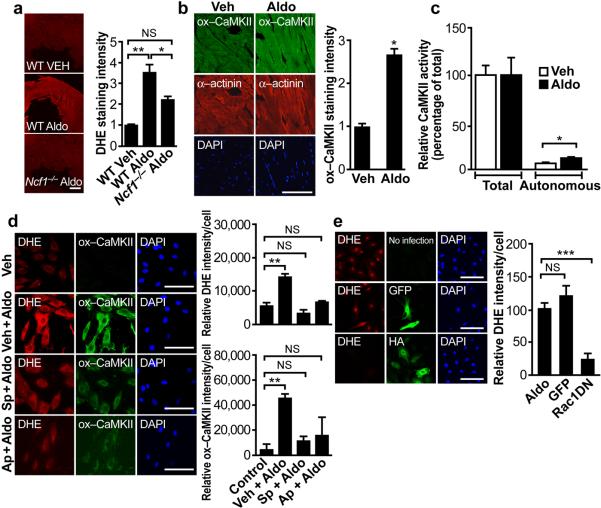
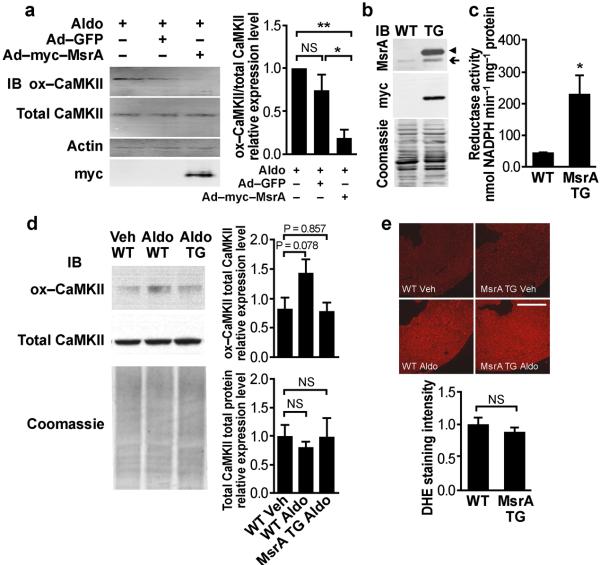
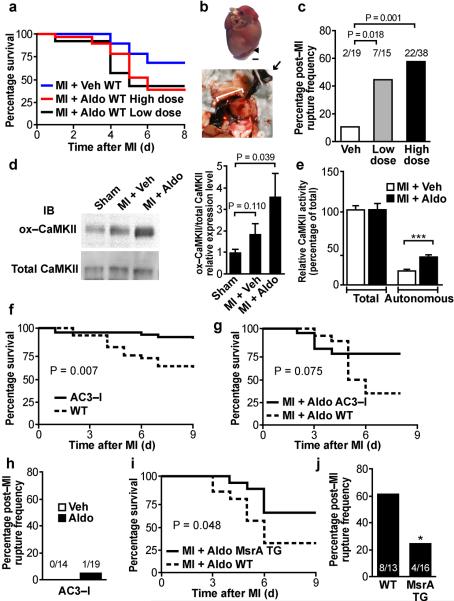
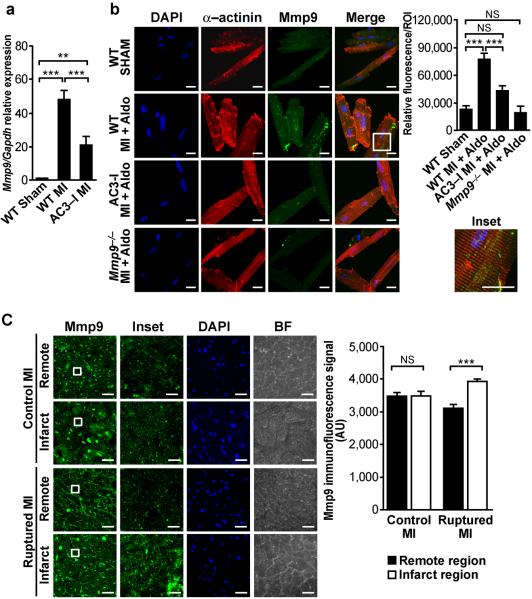
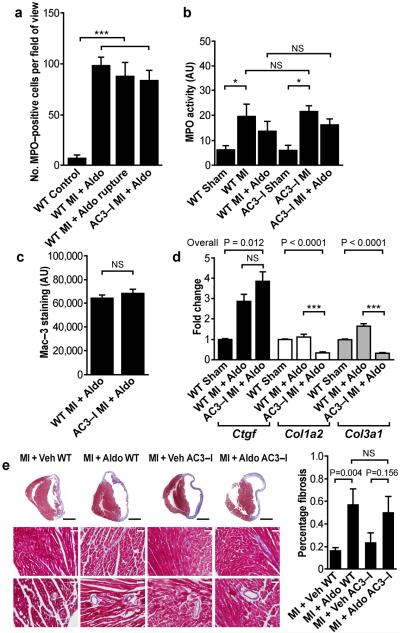
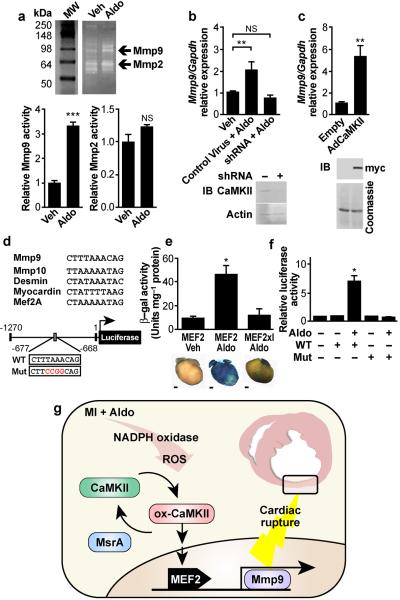
Excessive activation of the β-adrenergic, angiotensin II (Ang II) and aldosterone signaling pathways promotes mortality after myocardial infarction, and antagonists targeting these pathways are core therapies for treating this condition. Catecholamines and Ang II activate the multifunctional Ca(2+)/calmodulin-dependent protein kinase II (CaMKII), the inhibition of which prevents isoproterenol-mediated and Ang II-mediated cardiomyopathy. Here we show that aldosterone exerts direct toxic actions on myocardium by oxidative activation of CaMKII, causing cardiac rupture and increased mortality in mice after myocardial infarction. Aldosterone induces CaMKII oxidation by recruiting NADPH oxidase, and this oxidized and activated CaMKII promotes matrix metalloproteinase 9 (MMP9) expression in cardiomyocytes. Myocardial CaMKII inhibition, overexpression of methionine sulfoxide reductase A (an enzyme that reduces oxidized CaMKII) or NADPH oxidase deficiency prevented aldosterone-enhanced cardiac rupture after myocardial infarction. These findings show that oxidized myocardial CaMKII mediates the cardiotoxic effects of aldosterone on the cardiac matrix and establish CaMKII as a nodal signal for the neurohumoral pathways associated with poor outcomes after myocardial infarction.
Figures
References
-
- Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet. 2006;367:1747–1757. - PubMed
-
- Zhang R, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nature medicine. 2005;11:409–417. - PubMed
-
- Gutierrez-Marcos FM, et al. Atrial natriuretic peptide in patients with acute myocardial infarction without functional heart failure. Eur Heart J. 1991;12:503–507. - PubMed
-
- Beygui F, et al. High plasma aldosterone levels on admission are associated with death in patients presenting with acute ST-elevation myocardial infarction. Circulation. 2006;114:2604–2610. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01HL079031/HL/NHLBI NIH HHS/United States
- R01CA133114/CA/NCI NIH HHS/United States
- R01 HL096652/HL/NHLBI NIH HHS/United States
- P30 CA086862/CA/NCI NIH HHS/United States
- R01 HL061446/HL/NHLBI NIH HHS/United States
- R01 HL070250/HL/NHLBI NIH HHS/United States
- 1F30HL-095325/HL/NHLBI NIH HHS/United States
- F30 HL095325/HL/NHLBI NIH HHS/United States
- K26 RR017369/RR/NCRR NIH HHS/United States
- R01HL083422/HL/NHLBI NIH HHS/United States
- T32 GM067795/GM/NIGMS NIH HHS/United States
- R01 HL079031/HL/NHLBI NIH HHS/United States
- R01 HL083422/HL/NHLBI NIH HHS/United States
- R01HL096652/HL/NHLBI NIH HHS/United States
- T32 HL007121/HL/NHLBI NIH HHS/United States
- R01 CA133114/CA/NCI NIH HHS/United States
- RR-017369/RR/NCRR NIH HHS/United States
- R01HL70250/HL/NHLBI NIH HHS/United States
- T32 GM007337/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
