

Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes
- PMID: 22081229
- PMCID: PMC3245322
- DOI: 10.1038/ng.997
Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes
Abstract
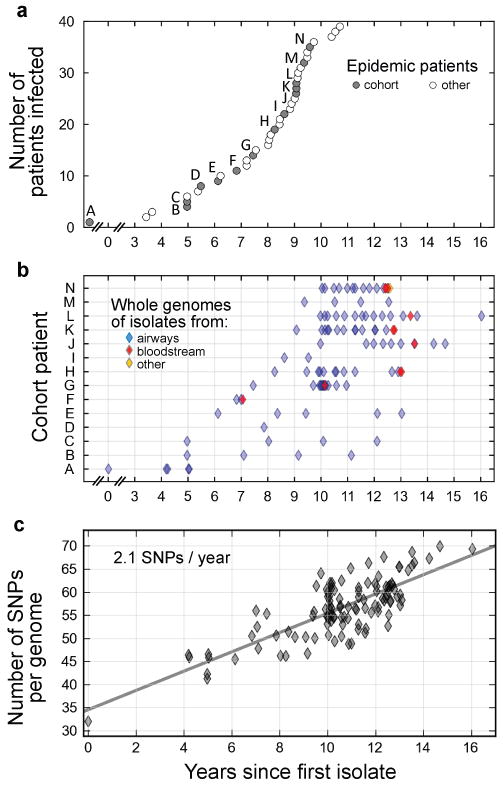
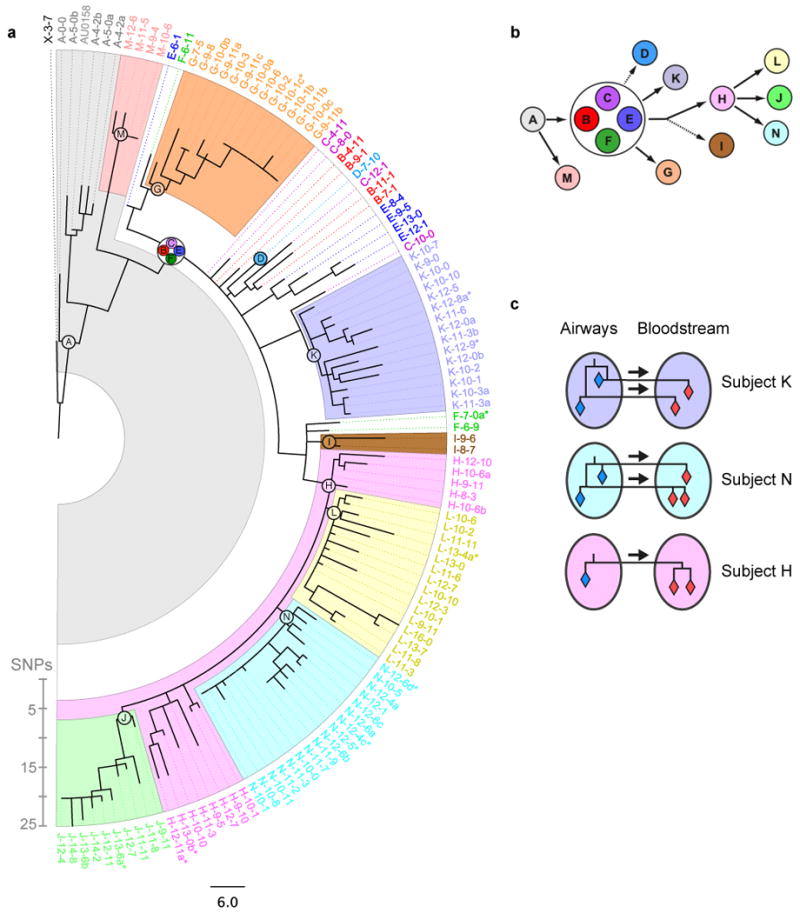
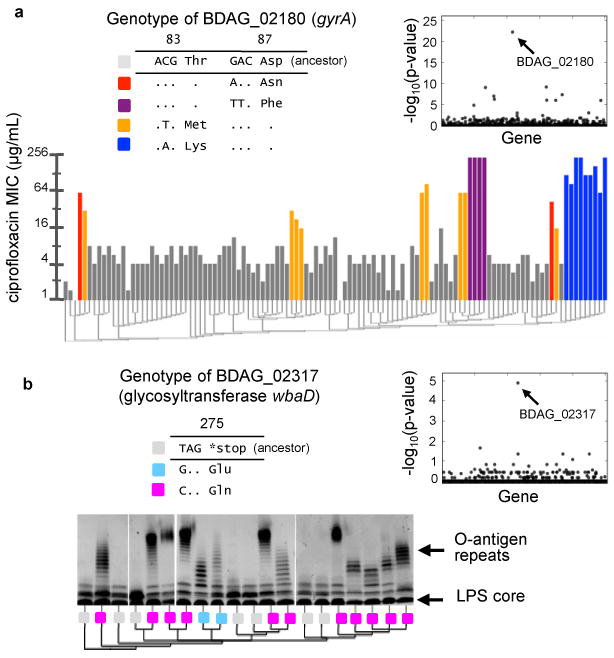
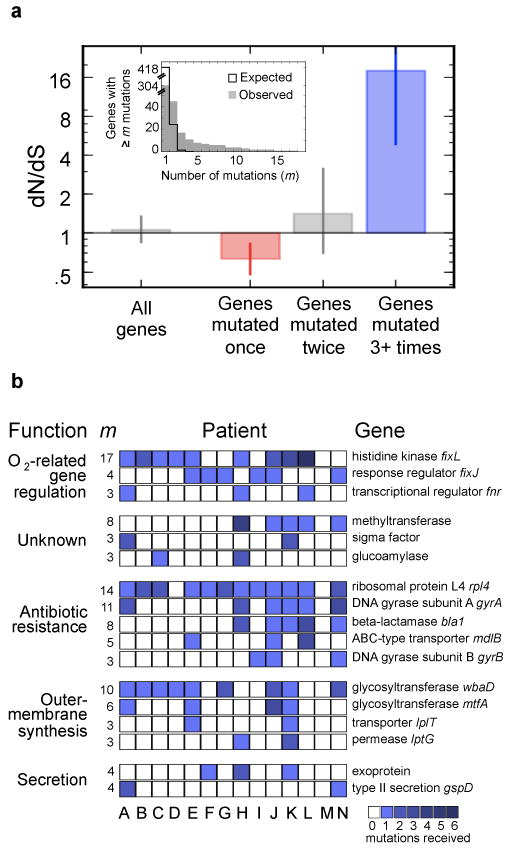
Bacterial pathogens evolve during the infection of their human host(1-8), but separating adaptive and neutral mutations remains challenging(9-11). Here we identify bacterial genes under adaptive evolution by tracking recurrent patterns of mutations in the same pathogenic strain during the infection of multiple individuals. We conducted a retrospective study of a Burkholderia dolosa outbreak among subjects with cystic fibrosis, sequencing the genomes of 112 isolates collected from 14 individuals over 16 years. We find that 17 bacterial genes acquired nonsynonymous mutations in multiple individuals, which indicates parallel adaptive evolution. Mutations in these genes affect important pathogenic phenotypes, including antibiotic resistance and bacterial membrane composition and implicate oxygen-dependent regulation as paramount in lung infections. Several genes have not previously been implicated in pathogenesis and may represent new therapeutic targets. The identification of parallel molecular evolution as a pathogen spreads among multiple individuals points to the key selection forces it experiences within human hosts.
Figures
Comment in
-
Chance and necessity in the evolution of a bacterial pathogen.Nat Genet. 2011 Nov 28;43(12):1174-6. doi: 10.1038/ng.1011. Nat Genet. 2011. PMID: 22120052
-
Bacterial evolution: Parallel lives.Nat Rev Microbiol. 2011 Dec 16;10(1):5. doi: 10.1038/nrmicro2722. Nat Rev Microbiol. 2011. PMID: 22173557 No abstract available.
-
Bacterial evolution: parallel lives.Nat Rev Genet. 2011 Dec 16;13(1):4. doi: 10.1038/nrg3135. Nat Rev Genet. 2011. PMID: 22179715 No abstract available.
References
-
- Suerbaum S, Josenhans C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat Rev Microbiol. 2007;5:441–452. - PubMed
-
- Musher DM, et al. Emergence of macrolide resistance during treatment of pneumococcal pneumonia. N Engl J Med. 2002;346:630–631. - PubMed
-
- Wong A, Kassen R. Parallel evolution and local differentiation in quinolone resistance in Pseudomonas aeruginosa. Microbiology. 2011;157:937–944. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
