

Characterising chromosome rearrangements: recent technical advances in molecular cytogenetics
- PMID: 22086080
- PMCID: PMC3238113
- DOI: 10.1038/hdy.2011.100
Characterising chromosome rearrangements: recent technical advances in molecular cytogenetics
Abstract
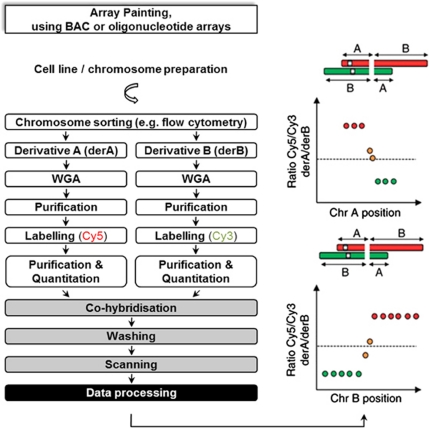
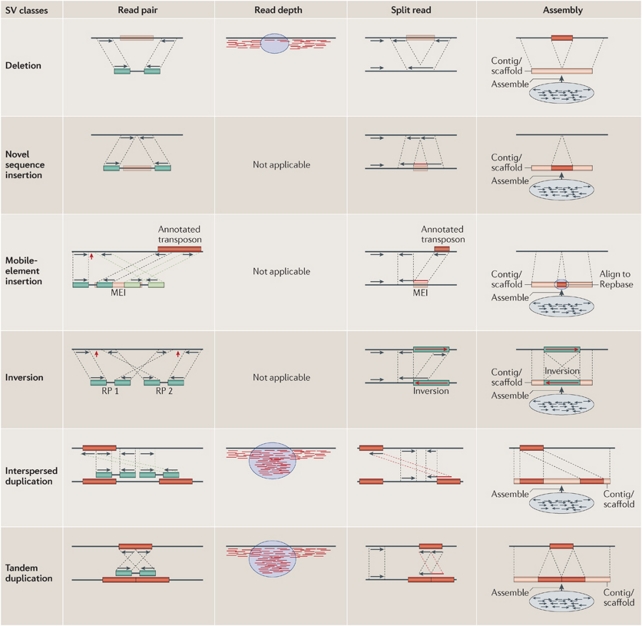
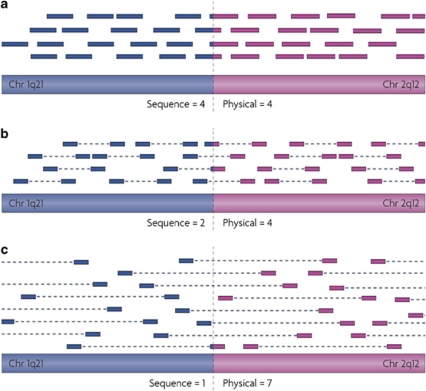
Genomic rearrangements can result in losses, amplifications, translocations and inversions of DNA fragments thereby modifying genome architecture, and potentially having clinical consequences. Many genomic disorders caused by structural variation have initially been uncovered by early cytogenetic methods. The last decade has seen significant progression in molecular cytogenetic techniques, allowing rapid and precise detection of structural rearrangements on a whole-genome scale. The high resolution attainable with these recently developed techniques has also uncovered the role of structural variants in normal genetic variation alongside single-nucleotide polymorphisms (SNPs). We describe how array-based comparative genomic hybridisation, SNP arrays, array painting and next-generation sequencing analytical methods (read depth, read pair and split read) allow the extensive characterisation of chromosome rearrangements in human genomes.
Figures


References
-
- Abel HJ, Duncavage EJ, Becker N, Armstrong JR, Magrini VJ, Pfeifer JD. SLOPE: a quick and accurate method for locating non-SNP structural variation from targeted next-generation sequence data. Bioinformatics. 2010;26:2684–2688. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
