

Pathophysiological insights in sickle cell disease
- PMID: 22089617
- PMCID: PMC3237253
Pathophysiological insights in sickle cell disease
Abstract
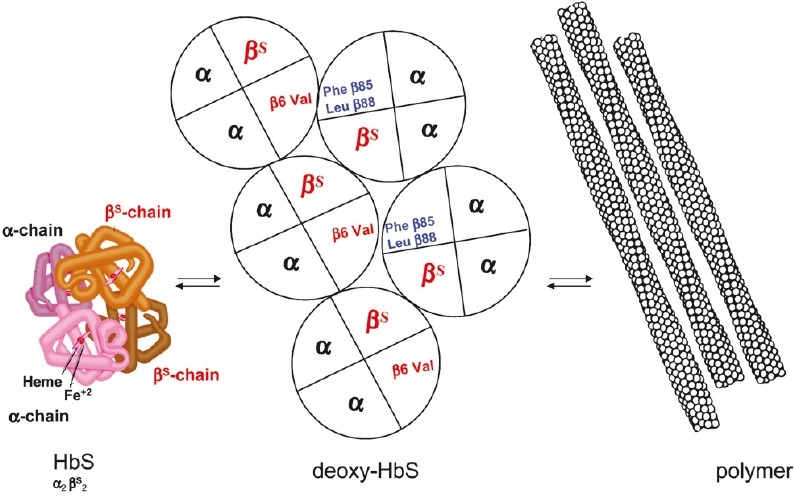
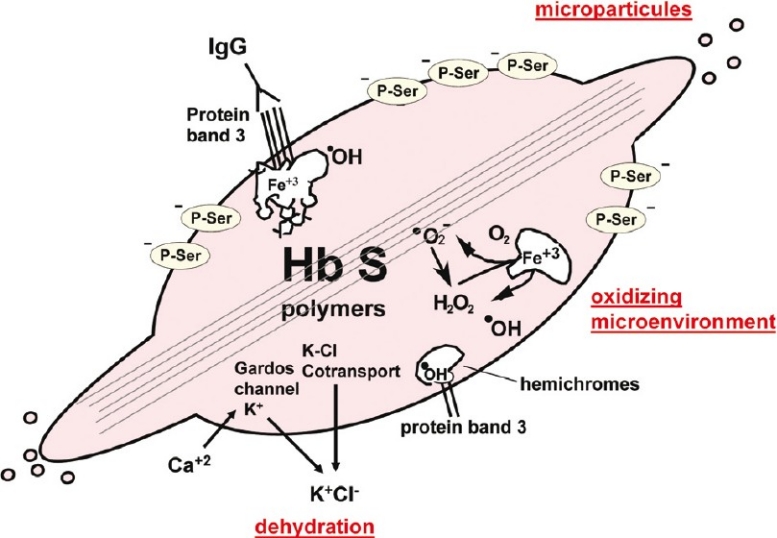
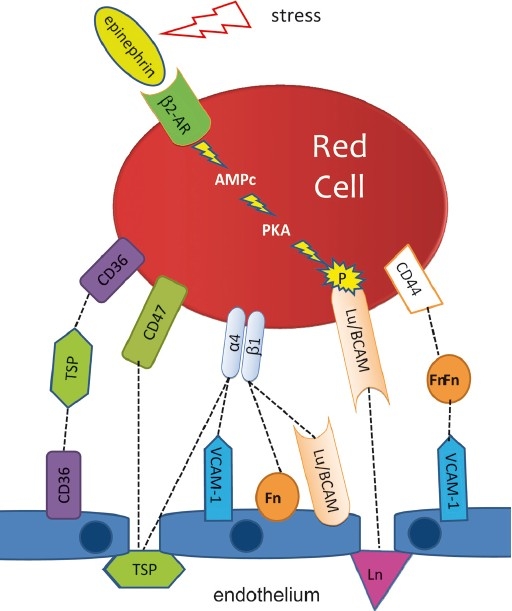
The first coherent pathophysiological scheme for sickle cell disease (SCD) emerged in the sixties-seventies based on an extremely detailed description of the molecular mechanism by which HbS in its deoxy-form polymerises and forms long fibres within the red blood cell that deform it and make it fragile. This scheme explains the haemolytic anaemia, and the mechanistic aspects of the vaso-occlusive crises (VOCs), but, even though it constitutes the basic mechanism of the disease, it does not account for the processes that actually trigger VOCs. This paper reviews recent data which imply: red blood cell dehydration, its abnormal adhesion properties to the endothelium, the participation of inflammatory phenomenon and of a global activation of all the cells present in the vessel, and finally, abnormalities of the vascular tone and of nitric oxide metabolism. These data altogether have shed a new light on the pathophysiology of the first molecular disease i.e. sickle cell disease.
Figures
References
-
- Pauling L, Itano H, Singer SJ, Wells IC. Sickle cell anemia: a molecular disease. Science. 1949;110:543–8. - PubMed
-
- Ingram VM. A specific chemical difference between the globins of normal human and sickle-cell anemia haemoglobin. Nature. 1956;178:792–4. - PubMed
-
- Bunn H, Forget BG. Hemoglobin: Molecular, genetic and clinical aspects. Philadelphia, PA, USA: WB Saunders; 1986.
-
- Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762–9. - PubMed
-
- Hebbel RP. Adhesive interactions of sickle erythrocytes with endothelium. J Clin Invest. 1997;100(11 Suppl):S83–6. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical