

Mitochondria in motor nerve terminals: function in health and in mutant superoxide dismutase 1 mouse models of familial ALS
- PMID: 22089637
- PMCID: PMC3237816
- DOI: 10.1007/s10863-011-9392-1
Mitochondria in motor nerve terminals: function in health and in mutant superoxide dismutase 1 mouse models of familial ALS
Abstract
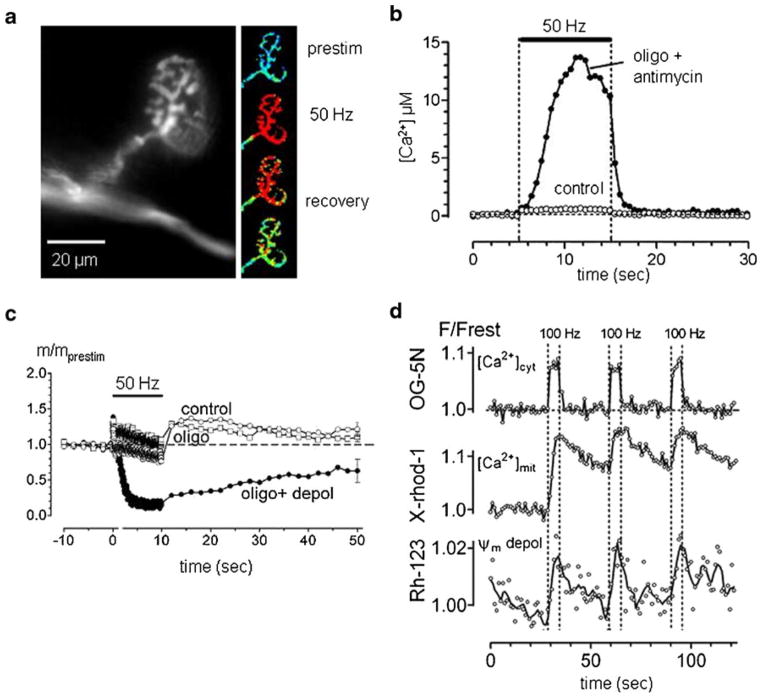
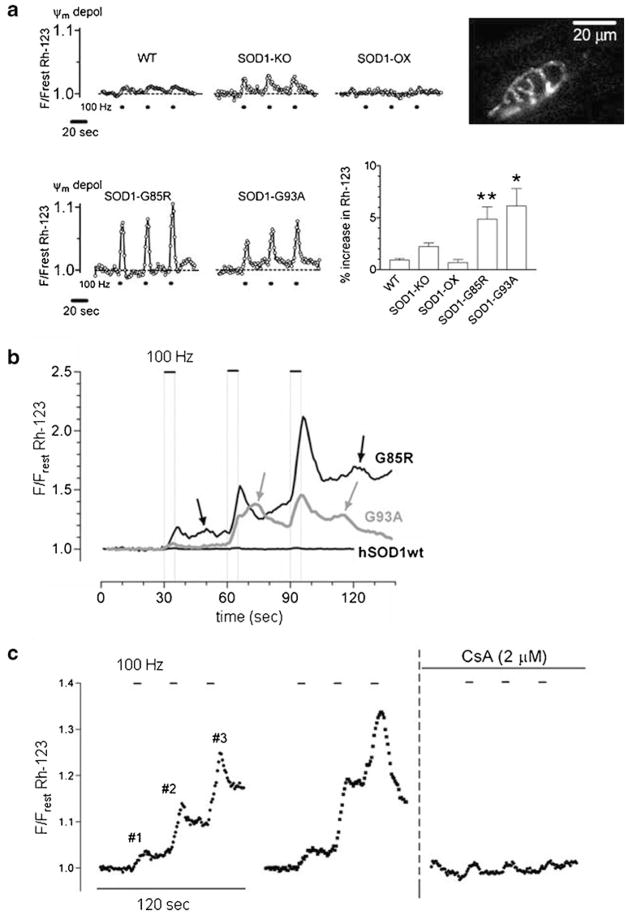
Mitochondria contribute to neuronal function not only via their ability to generate ATP, but also via their ability to buffer large Ca(2+) loads. This review summarizes evidence that mitochondrial Ca(2+) sequestration is especially important for sustaining the function of vertebrate motor nerve terminals during repetitive stimulation. Motor terminal mitochondria can sequester large amounts of Ca(2+) because they have mechanisms for limiting both the mitochondrial depolarization and the increase in matrix free [Ca(2+)] associated with Ca(2+) influx. In mice expressing mutations of human superoxide dismutase -1 (SOD1) that cause some cases of familial amyotrophic lateral sclerosis (fALS), motor terminals degenerate well before the death of motor neuron cell bodies. This review presents evidence for early and progressive mitochondrial dysfunction in motor terminals of mutant SOD1 mice (G93A, G85R). This dysfunction would impair mitochondrial ability to sequester stimulation-associated Ca(2+) loads, and thus likely contributes to the early degeneration of motor terminals.
Figures
Similar articles
-
Mitochondrial dysfunction in familial amyotrophic lateral sclerosis.J Bioenerg Biomembr. 2011 Dec;43(6):587-92. doi: 10.1007/s10863-011-9393-0. J Bioenerg Biomembr. 2011. PMID: 22072073 Review.
-
Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex.J Biol Chem. 2007 Jun 1;282(22):16691-9. doi: 10.1074/jbc.M609743200. Epub 2007 Apr 2. J Biol Chem. 2007. PMID: 17403682
-
Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1.Neurobiol Dis. 2000 Dec;7(6 Pt B):623-43. doi: 10.1006/nbdi.2000.0299. Neurobiol Dis. 2000. PMID: 11114261
-
Repetitive nerve stimulation transiently opens the mitochondrial permeability transition pore in motor nerve terminals of symptomatic mutant SOD1 mice.Neurobiol Dis. 2011 Jun;42(3):381-90. doi: 10.1016/j.nbd.2011.01.031. Epub 2011 Feb 18. Neurobiol Dis. 2011. PMID: 21310237 Free PMC article.
-
Transgenic mice with human mutant genes causing Parkinson's disease and amyotrophic lateral sclerosis provide common insight into mechanisms of motor neuron selective vulnerability to degeneration.Rev Neurosci. 2007;18(2):115-36. doi: 10.1515/revneuro.2007.18.2.115. Rev Neurosci. 2007. PMID: 17593875 Review.
Cited by
-
Structure and Function of the Mammalian Neuromuscular Junction.Compr Physiol. 2022 Aug 11;12(4):3731-3766. doi: 10.1002/cphy.c210022. Compr Physiol. 2022. PMID: 35950651 Free PMC article.
-
Targeting mitochondrial Ca2+ uptake for the treatment of amyotrophic lateral sclerosis.J Physiol. 2024 Apr;602(8):1519-1549. doi: 10.1113/JP284143. Epub 2023 Nov 27. J Physiol. 2024. PMID: 38010626 Free PMC article. Review.
-
Purkinje neuron Ca2+ influx reduction rescues ataxia in SCA28 model.J Clin Invest. 2015 Jan;125(1):263-74. doi: 10.1172/JCI74770. Epub 2014 Dec 8. J Clin Invest. 2015. PMID: 25485680 Free PMC article.
-
SOD2 in mitochondrial dysfunction and neurodegeneration.Free Radic Biol Med. 2013 Sep;62:4-12. doi: 10.1016/j.freeradbiomed.2013.05.027. Epub 2013 May 29. Free Radic Biol Med. 2013. PMID: 23727323 Free PMC article. Review.
-
Elevated mRNA-levels of distinct mitochondrial and plasma membrane Ca(2+) transporters in individual hypoglossal motor neurons of endstage SOD1 transgenic mice.Front Cell Neurosci. 2014 Nov 14;8:353. doi: 10.3389/fncel.2014.00353. eCollection 2014. Front Cell Neurosci. 2014. PMID: 25452714 Free PMC article.
References
-
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jemkins NA, Copeland NG, et al. Neuron. 1997;18:327–338. - PubMed
-
- Burke RE. In: Myology. 3. Engel AG, Franzini-Armstrong D, editors. McGraw-Hill; New York: 2004.
-
- Chiu AY, Zhai P, Dalcanto MC, Peters TM, Kwon YW, Prattis SM, et al. Mol Cell Neurosci. 1995;6:349–362. - PubMed
-
- Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, et al. J Neurochem. 2006;96:1349–1361. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
