

Functional genomic analysis unravels a metabolic-inflammatory interplay in adrenoleukodystrophy
- PMID: 22095690
- PMCID: PMC3277307
- DOI: 10.1093/hmg/ddr536
Functional genomic analysis unravels a metabolic-inflammatory interplay in adrenoleukodystrophy
Abstract
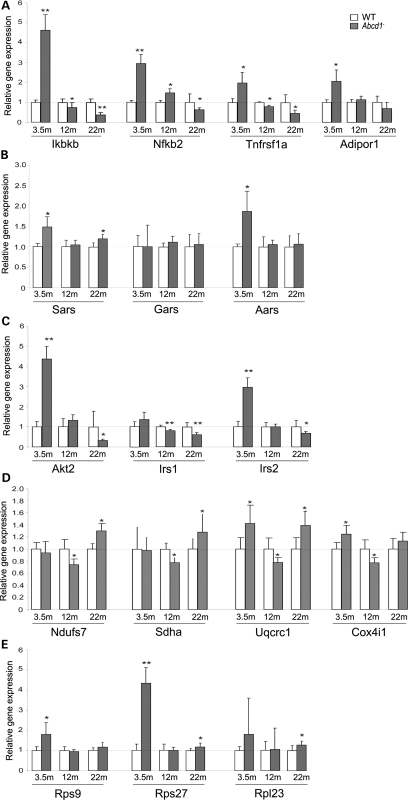
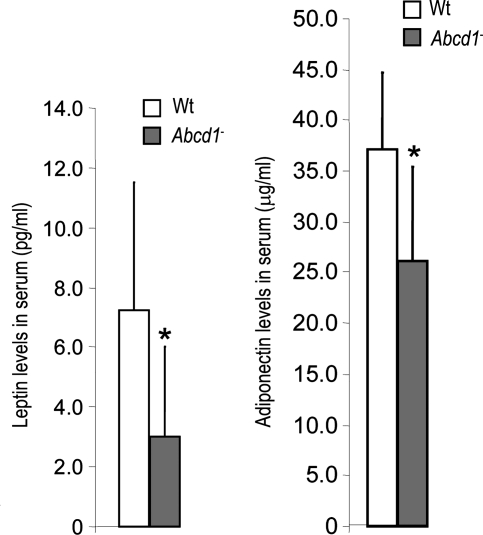
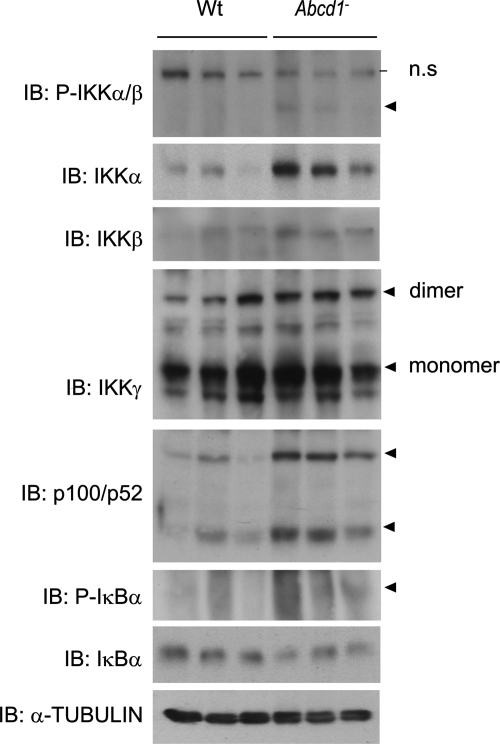
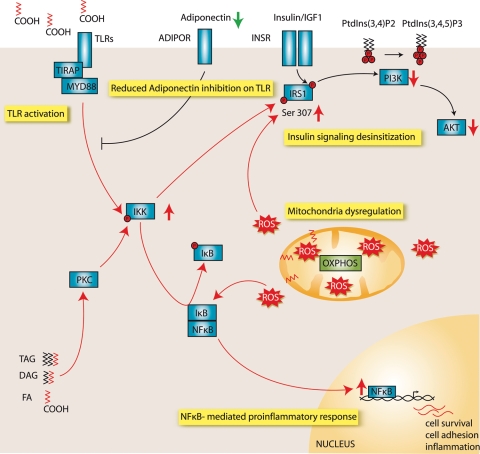
X-linked adrenoleukodystrophy (X-ALD) is an inherited disorder characterized by axonopathy and demyelination in the central nervous system and adrenal insufficiency. Main X-ALD phenotypes are: (i) an adult adrenomyeloneuropathy (AMN) with axonopathy in spinal cords, (ii) cerebral AMN with brain demyelination (cAMN) and (iii) a childhood variant, cALD, characterized by severe cerebral demyelination. Loss of function of the ABCD1 peroxisomal fatty acid transporter and subsequent accumulation of very-long-chain fatty acids (VLCFAs) are the common culprits to all forms of X-ALD, an aberrant microglial activation accounts for the cerebral forms, whereas inflammation allegedly plays no role in AMN. How VLCFA accumulation leads to neurodegeneration and what factors account for the dissimilar clinical outcomes and prognosis of X-ALD variants remain elusive. To gain insights into these questions, we undertook a transcriptomic approach followed by a functional-enrichment analysis in spinal cords of the animal model of AMN, the Abcd1(-) null mice, and in normal-appearing white matter of cAMN and cALD patients. We report that the mouse model shares with cAMN and cALD a common signature comprising dysregulation of oxidative phosphorylation, adipocytokine and insulin signaling pathways, and protein synthesis. Functional validation by quantitative polymerase chain reaction, western blots and assays in spinal cord organotypic cultures confirmed the interplay of these pathways through IkB kinase, being VLCFA in excess a causal, upstream trigger promoting the altered signature. We conclude that X-ALD is, in all its variants, a metabolic/inflammatory syndrome, which may offer new targets in X-ALD therapeutics.
Figures





References
-
- Ferrer I., Aubourg P., Pujol A. General aspects and neuropathology of X-linked adrenoleukodystrophy. Brain Pathol. 2010;20:817–830. doi:10.1111/j.1750-3639.2010.00390.x. - DOI - PMC - PubMed
-
- Mosser J., Douar A.M., Sarde C.O., Kioschis P., Feil R., Moser H., Poustka A.M., Mandel J.L., Aubourg P. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature. 1993;361:726–730. doi:10.1038/361726a0. - DOI - PubMed
-
- van Roermund C.W., Visser W.F., Ijlst L., van Cruchten A., Boek M., Kulik W., Waterham H.R., Wanders R.J. The human peroxisomal ABC half transporter ALDP functions as a homodimer and accepts acyl-CoA esters. FASEB J. 2008;22:4201–4208. doi:10.1096/fj.08-110866. - DOI - PubMed
-
- Kemp S., Pujol A., Waterham H.R., van Geel B.M., Boehm C.D., Raymond G.V., Cutting G.R., Wanders R.J., Moser H.W. ABCD1 mutations and the X-linked adrenoleukodystrophy mutation database: role in diagnosis and clinical correlations. Hum. Mutat. 2001;18:499–515. doi:10.1002/humu.1227. - DOI - PubMed
-
- Moser H., Smith K.D., Watkins P.A., Powers J., Moser A.B. In: The Metabolic and Molecular Bases of Inherited Disease. Scriver C., editor. New York: McGraw-Hill; 2001. pp. 3257–3301.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
