

Regulation of peroxisome proliferator-activated receptor-γ by angiotensin II via transforming growth factor-β1-activated p38 mitogen-activated protein kinase in aortic smooth muscle cells
- PMID: 22095985
- PMCID: PMC3262055
- DOI: 10.1161/ATVBAHA.111.239897
Regulation of peroxisome proliferator-activated receptor-γ by angiotensin II via transforming growth factor-β1-activated p38 mitogen-activated protein kinase in aortic smooth muscle cells
Abstract
Objective: Peroxisome proliferator-activated receptor-γ (PPARγ) ligands attenuate angiotensin II (Ang II)-induced atherosclerosis through interactions with vascular smooth muscle cell (VSMC)-specific PPARγ in hypercholesterolemic mice. Therefore, the purpose of this study was to determine the mechanism of Ang II-mediated intracellular regulation of PPARγ in VSMCs.
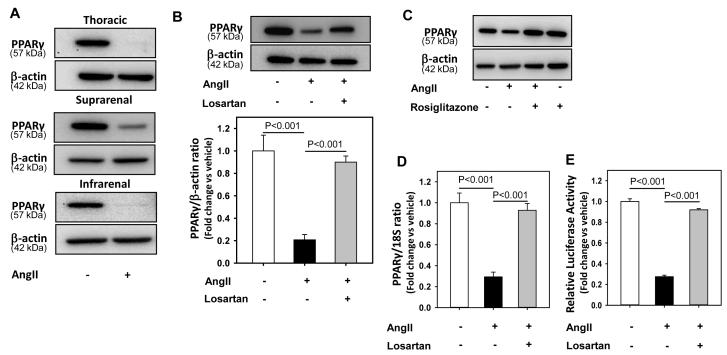
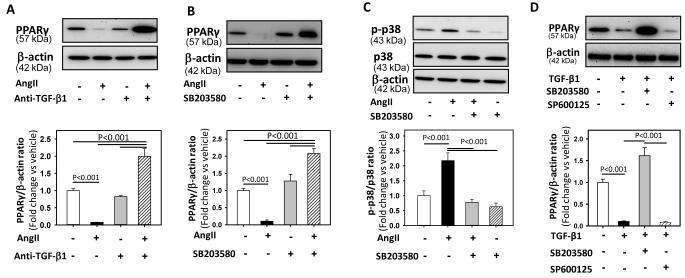
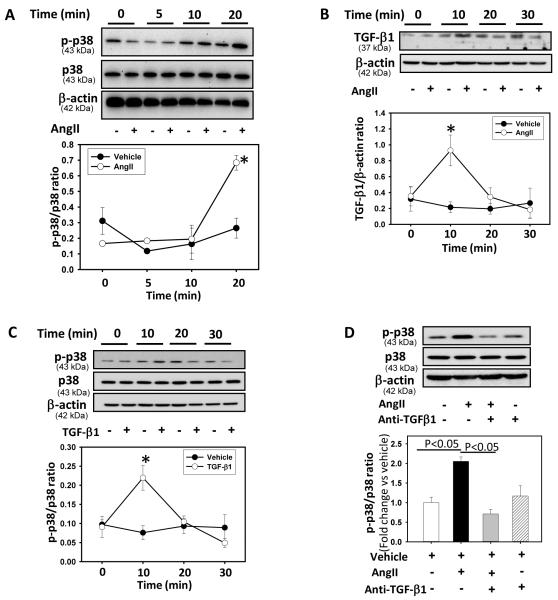
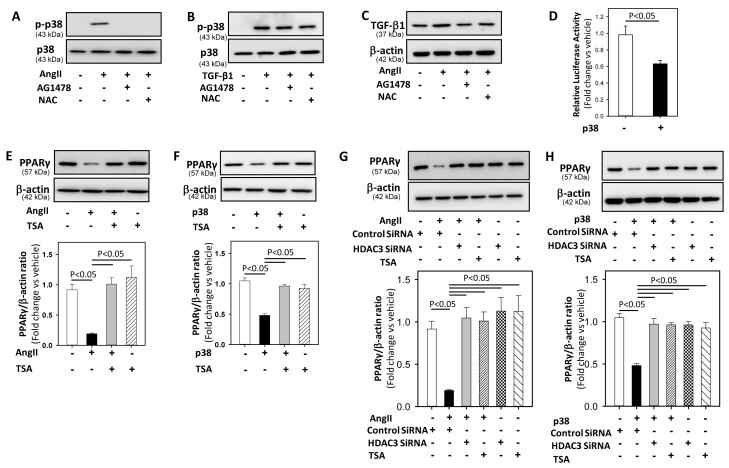
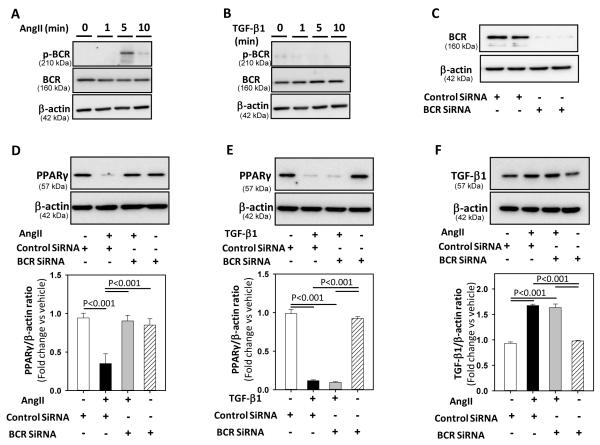
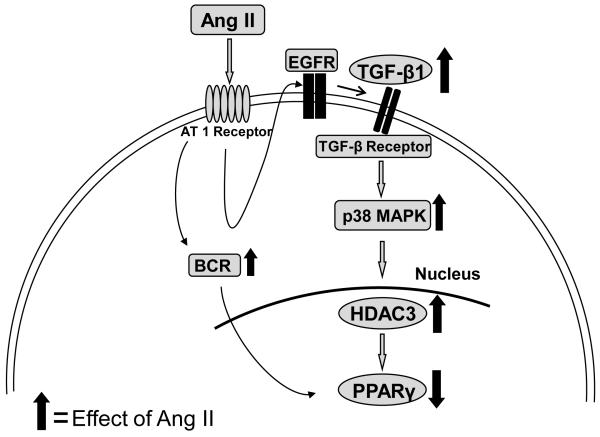
Methods and results: Incubation of cultured mouse aortic VSMCs with Ang II for 24 hours reduced abundance of PPARγ protein, mRNA, and transcriptional activity (P<0.001). This effect was attenuated by an angiotensin type 1 receptor antagonist, losartan. Ang II-induced PPARγ reduction was dependent on stimulation of transforming growth factor (TGF)-β1 as demonstrated using either a neutralizing antibody or small interfering RNA (siRNA). Ang II-induced TGF-β1 secretion was dependent on epidermal growth factor receptor kinase activation through reactive oxygen species production. Inhibition of p38 mitogen-activated protein kinase by SB203580 or siRNA inhibited both Ang II- and TGF-β1-induced PPARγ reduction. Blockade of TGF-β1 decreased p38 phosphorylation induced by Ang II. siRNA-mediated inhibition of histone deacetylase 3 attenuated p38-mediated reductions in PPARγ abundance.
Conclusions: These findings suggest that Ang II decreases PPARγ abundance in cultured VSMCs via an angiotensin type 1 receptor-dependent secretion of TGF-β1 via phosphorylation of p38 mitogen-activated protein kinase and histone deacetylase 3.
Figures
References
-
- Schoonjans K, Martin G, Staels B, Auwerx J. Peroxisome proliferator-activated receptors, orphans with ligands and functions. Current Opinion in Lipidol. 1997;8:159–166. - PubMed
-
- Hsueh WA, Bruemmer D. Peroxisome proliferator-activated receptor gamma: implications for cardiovascular disease. Hypertension. 2004;43:297–305. - PubMed
-
- Levi Z, Shaish A, Yacov N, Levkovitz H, Trestman S, Gerber Y, Cohen H, Dvir A, Rhachmani R, Ravid M, Harats D. Rosiglitazone (PPARgamma-agonist) attenuates atherogenesis with no effect on hyperglycaemia in a combined diabetes-atherosclerosis mouse model. Diabetes Obes Metab. 2003;5:45–50. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
