

Whole-genome comparison of two Campylobacter jejuni isolates of the same sequence type reveals multiple loci of different ancestral lineage
- PMID: 22096527
- PMCID: PMC3214069
- DOI: 10.1371/journal.pone.0027121
Whole-genome comparison of two Campylobacter jejuni isolates of the same sequence type reveals multiple loci of different ancestral lineage
Abstract
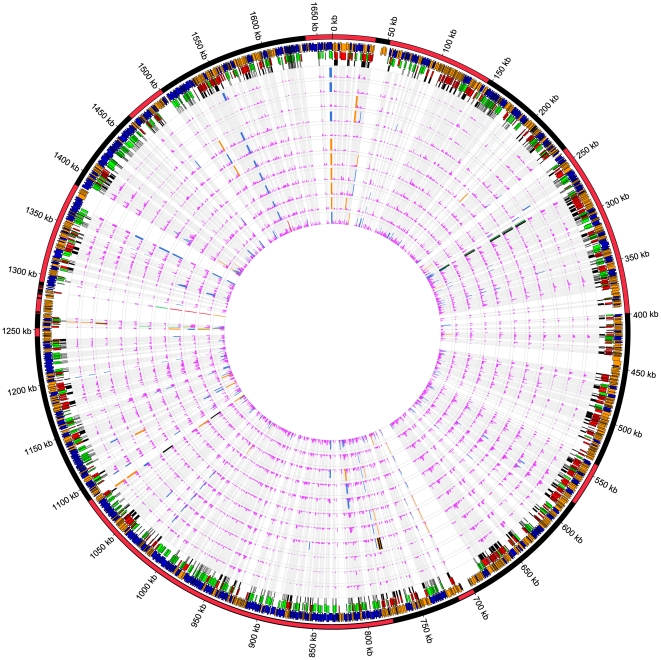
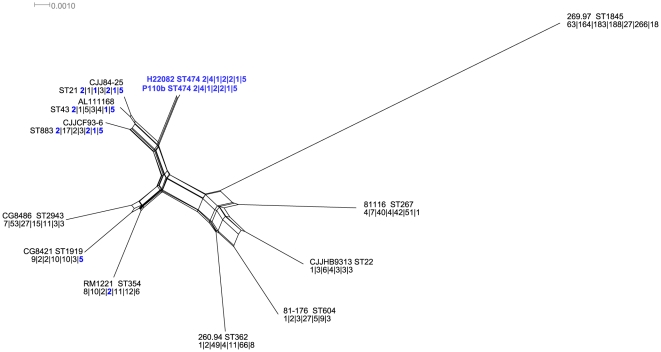
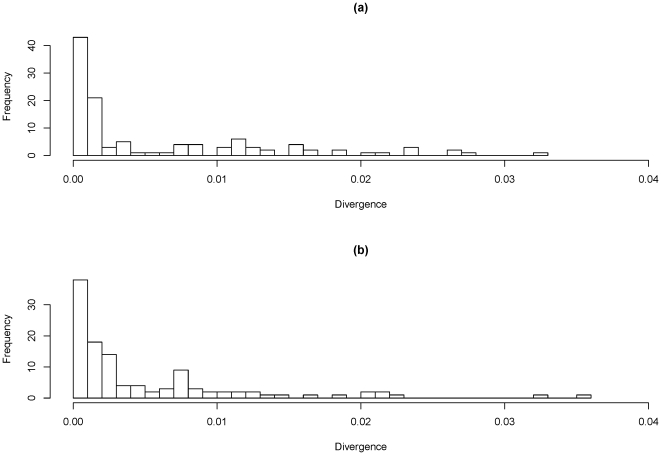
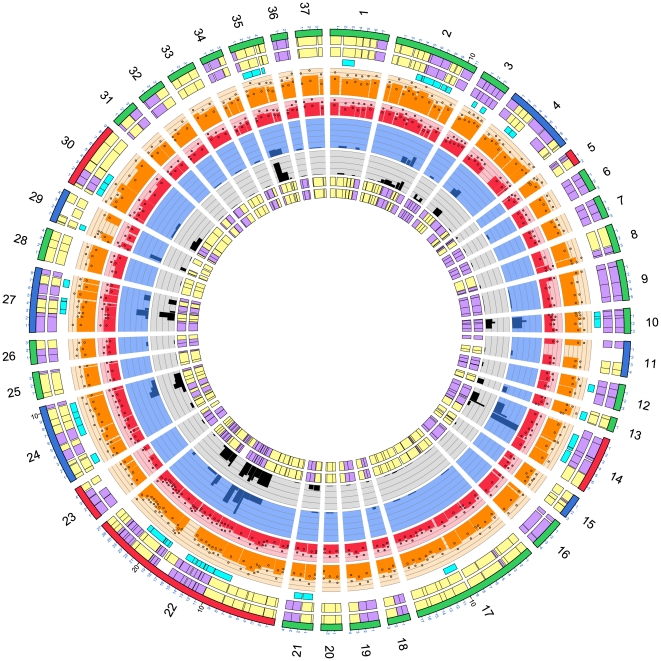
Campylobacter jejuni ST-474 is the most important human enteric pathogen in New Zealand, and yet this genotype is rarely found elsewhere in the world. Insight into the evolution of this organism was gained by a whole genome comparison of two ST-474, flaA SVR-14 isolates and other available C. jejuni isolates and genomes. The two isolates were collected from different sources, human (H22082) and retail poultry (P110b), at the same time and from the same geographical location. Solexa sequencing of each isolate resulted in ~1.659 Mb (H22082) and ~1.656 Mb (P110b) of assembled sequences within 28 (H22082) and 29 (P110b) contigs. We analysed 1502 genes for which we had sequences within both ST-474 isolates and within at least one of 11 C. jejuni reference genomes. Although 94.5% of genes were identical between the two ST-474 isolates, we identified 83 genes that differed by at least one nucleotide, including 55 genes with non-synonymous substitutions. These covered 101 kb and contained 672 point differences. We inferred that 22 (3.3%) of these differences were due to mutation and 650 (96.7%) were imported via recombination. Our analysis estimated 38 recombinant breakpoints within these 83 genes, which correspond to recombination events affecting at least 19 loci regions and gives a tract length estimate of ~2 kb. This includes a ~12 kb region displaying non-homologous recombination in one of the ST-474 genomes, with the insertion of two genes, including ykgC, a putative oxidoreductase, and a conserved hypothetical protein of unknown function. Furthermore, our analysis indicates that the source of this recombined DNA is more likely to have come from C. jejuni strains that are more closely related to ST-474. This suggests that the rates of recombination and mutation are similar in order of magnitude, but that recombination has been much more important for generating divergence between the two ST-474 isolates.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
