

Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization
- PMID: 22098752
- PMCID: PMC3218324
- DOI: 10.1016/j.bpj.2011.10.024
Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization
Abstract
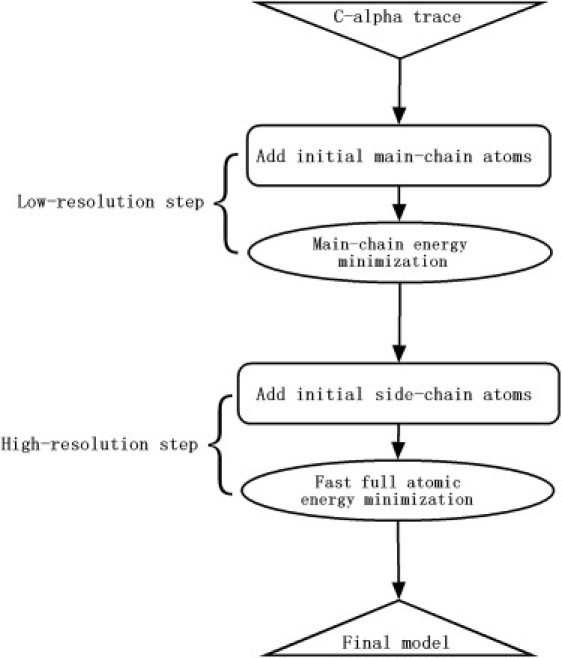
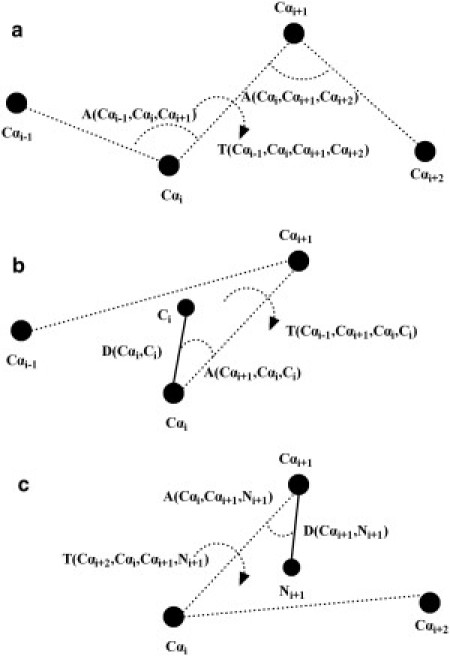
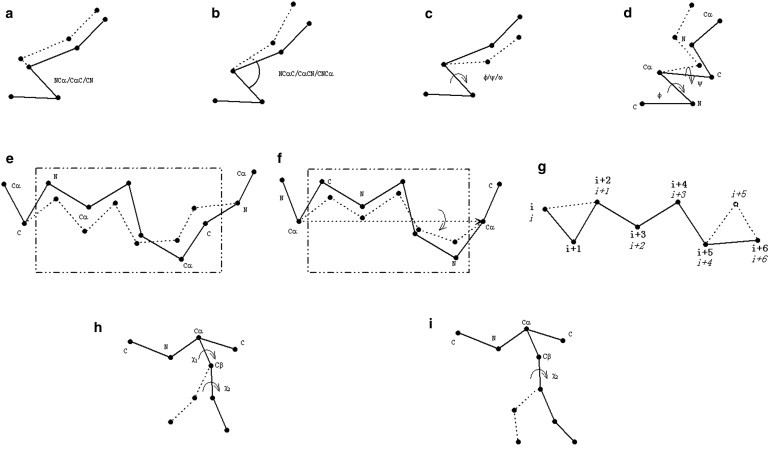
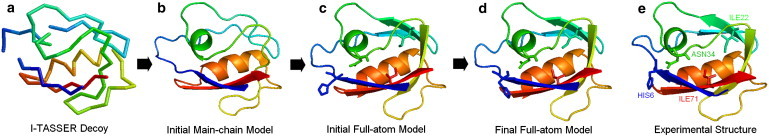
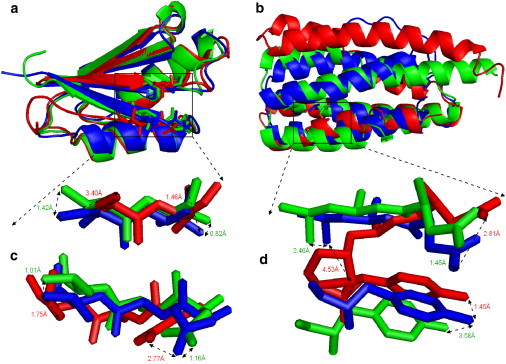
Most protein structural prediction algorithms assemble structures as reduced models that represent amino acids by a reduced number of atoms to speed up the conformational search. Building accurate full-atom models from these reduced models is a necessary step toward a detailed function analysis. However, it is difficult to ensure that the atomic models retain the desired global topology while maintaining a sound local atomic geometry because the reduced models often have unphysical local distortions. To address this issue, we developed a new program, called ModRefiner, to construct and refine protein structures from Cα traces based on a two-step, atomic-level energy minimization. The main-chain structures are first constructed from initial Cα traces and the side-chain rotamers are then refined together with the backbone atoms with the use of a composite physics- and knowledge-based force field. We tested the method by performing an atomic structure refinement of 261 proteins with the initial models constructed from both ab initio and template-based structure assemblies. Compared with other state-of-art programs, ModRefiner shows improvements in both global and local structures, which have more accurate side-chain positions, better hydrogen-bonding networks, and fewer atomic overlaps. ModRefiner is freely available at http://zhanglab.ccmb.med.umich.edu/ModRefiner.
Copyright © 2011 Biophysical Society. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Simons K.T., Ruczinski I., et al. Baker D. Improved recognition of native-like protein structures using a combination of sequence-dependent and sequence-independent features of proteins. Proteins. 1999;34:82–95. - PubMed
-
- Bradley P., Misura K.M., Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309:1868–1871. - PubMed
-
- Kabsch W. A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. 1976;32A:922–923.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
