

Inhibitors of membranous adenylyl cyclases
- PMID: 22100304
- PMCID: PMC3273670
- DOI: 10.1016/j.tips.2011.10.006
Inhibitors of membranous adenylyl cyclases
Abstract
Membranous adenylyl cyclases (mACs) constitute a family of nine isoforms with different expression patterns. Studies with mAC gene knockout mice provide evidence for the notion that AC isoforms play distinct (patho)physiological roles. Consequently, there is substantial interest in the development of isoform-selective mAC inhibitors. Here, we review the current literature on mAC inhibitors. Structurally diverse inhibitors targeting the catalytic site and allosteric sites (e.g. the diterpene site) have been identified. The catalytic site of mACs accommodates both purine and pyrimidine nucleotides, with a hydrophobic pocket constituting a major affinity-conferring domain for substituents at the 2'- and 3'-O-ribosyl position of nucleotides. BODIPY-forskolin stimulates ACs 1 and 5 but inhibits AC2. However, so far, no inhibitor has been examined at all mAC isoforms, and data obtained with mAC inhibitors in intact cells have not always been interpreted cautiously enough. Future strategies for the development of the mAC inhibitor field are discussed critically.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures






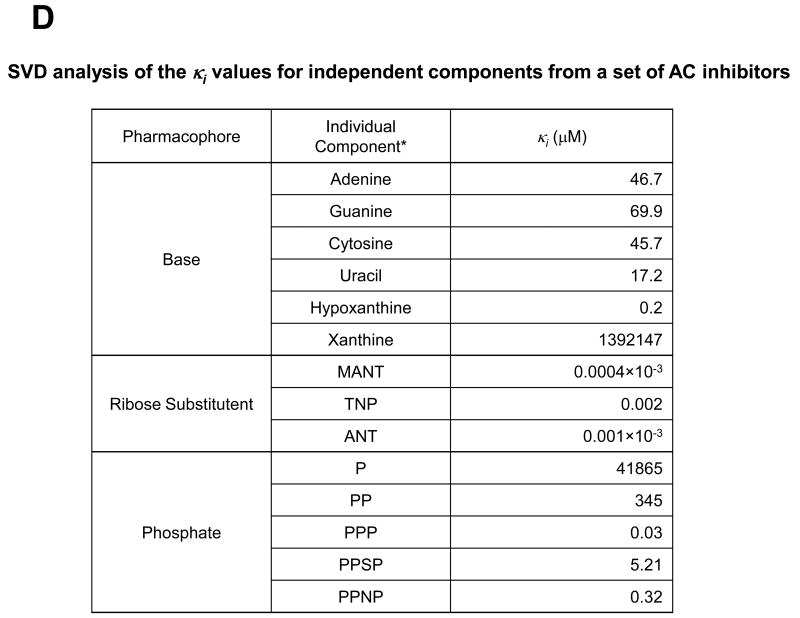
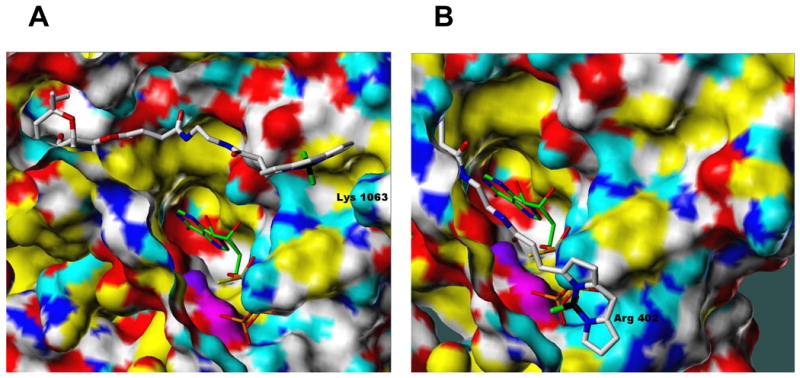

Similar articles
-
Differential inhibition of adenylyl cyclase isoforms and soluble guanylyl cyclase by purine and pyrimidine nucleotides.J Biol Chem. 2004 May 7;279(19):19955-69. doi: 10.1074/jbc.M312560200. Epub 2004 Feb 23. J Biol Chem. 2004. PMID: 14981084
-
International Union of Basic and Clinical Pharmacology. CI. Structures and Small Molecule Modulators of Mammalian Adenylyl Cyclases.Pharmacol Rev. 2017 Apr;69(2):93-139. doi: 10.1124/pr.116.013078. Pharmacol Rev. 2017. PMID: 28255005 Free PMC article. Review.
-
Broad specificity of mammalian adenylyl cyclase for interaction with 2',3'-substituted purine- and pyrimidine nucleotide inhibitors.Mol Pharmacol. 2006 Sep;70(3):878-86. doi: 10.1124/mol.106.026427. Epub 2006 Jun 9. Mol Pharmacol. 2006. PMID: 16766715
-
Differential inhibition of various adenylyl cyclase isoforms and soluble guanylyl cyclase by 2',3'-O-(2,4,6-trinitrophenyl)-substituted nucleoside 5'-triphosphates.J Pharmacol Exp Ther. 2009 Sep;330(3):687-95. doi: 10.1124/jpet.109.155432. Epub 2009 Jun 3. J Pharmacol Exp Ther. 2009. PMID: 19494187 Free PMC article.
-
Mammalian Nucleotidyl Cyclases and Their Nucleotide Binding Sites.Handb Exp Pharmacol. 2017;238:49-66. doi: 10.1007/164_2015_34. Handb Exp Pharmacol. 2017. PMID: 27900607 Review.
Cited by
-
The role of the type 7 adenylyl cyclase isoform in alcohol use disorder and depression.Front Pharmacol. 2022 Oct 28;13:1012013. doi: 10.3389/fphar.2022.1012013. eCollection 2022. Front Pharmacol. 2022. PMID: 36386206 Free PMC article. Review.
-
A door opener for future research: agonist-induced β3-adrenoceptor desensitization in HEK cells but not CHO cells.Naunyn Schmiedebergs Arch Pharmacol. 2013 Oct;386(10):841-2. doi: 10.1007/s00210-013-0884-x. Epub 2013 Jun 12. Naunyn Schmiedebergs Arch Pharmacol. 2013. PMID: 23756577 No abstract available.
-
MDL-12330A potentiates TRAIL-induced apoptosis in gastric cancer cells through CHOP-mediated DR5 upregulation.Korean J Physiol Pharmacol. 2017 Jul;21(4):397-405. doi: 10.4196/kjpp.2017.21.4.397. Epub 2017 Jun 26. Korean J Physiol Pharmacol. 2017. PMID: 28706453 Free PMC article.
-
Is cIMP a second messenger with functions opposite to those of cGMP?Naunyn Schmiedebergs Arch Pharmacol. 2014 Sep;387(9):897-9. doi: 10.1007/s00210-014-1013-1. Epub 2014 Jul 15. Naunyn Schmiedebergs Arch Pharmacol. 2014. PMID: 25017018 No abstract available.
-
Bithionol Potently Inhibits Human Soluble Adenylyl Cyclase through Binding to the Allosteric Activator Site.J Biol Chem. 2016 Apr 29;291(18):9776-84. doi: 10.1074/jbc.M115.708255. Epub 2016 Mar 9. J Biol Chem. 2016. PMID: 26961873 Free PMC article.
References
-
- Sunahara RK, Taussig R. Isoforms of mammalian adenylyl cyclase: multiplicities of signalling. Mol Interv. 2002;2:168–184. - PubMed
-
- Pierre S, et al. Capturing adenylyl cyclases as potential drug targets. Nat Rev Drug Discov. 2009;8:321–335. - PubMed
-
- Laurenza A, et al. Forskolin: a specific stimulator of adenylyl cyclase or a diterpene with multiple sites of action? Trends Pharmacol Sci. 1989;10:442–447. - PubMed
-
- Putnam W, et al. Identification of a forskolin-like molecule in human renal cysts. J Am Soc Nephrol. 2007;18:934–943. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
