

Novel role of p66Shc in ROS-dependent VEGF signaling and angiogenesis in endothelial cells
- PMID: 22101521
- PMCID: PMC3353779
- DOI: 10.1152/ajpheart.00739.2011
Novel role of p66Shc in ROS-dependent VEGF signaling and angiogenesis in endothelial cells
Abstract
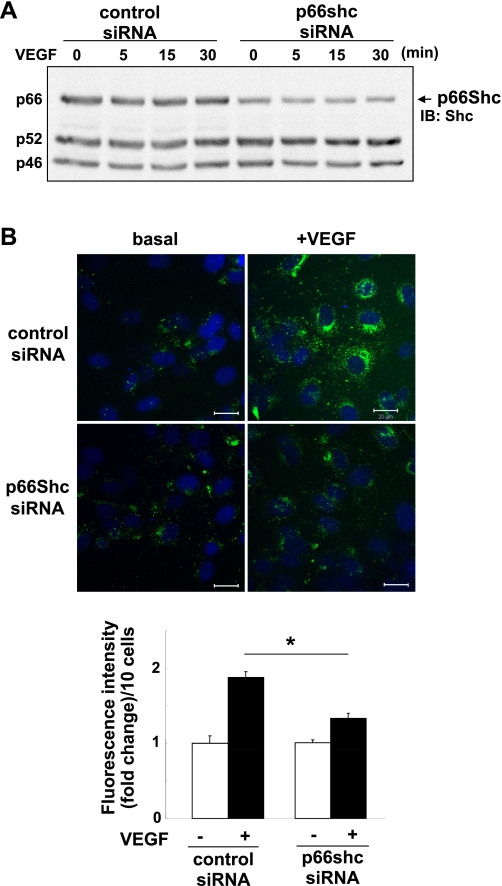
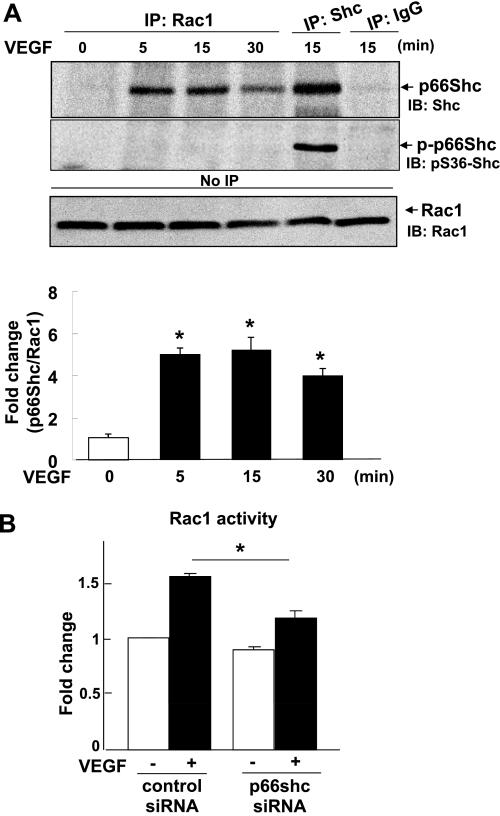
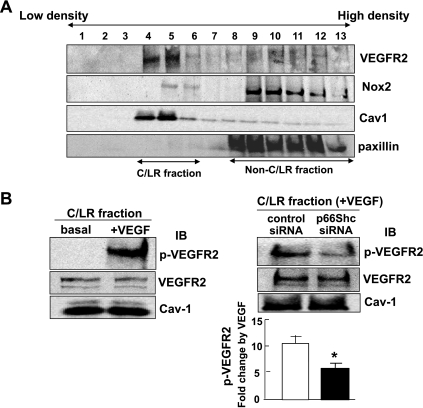
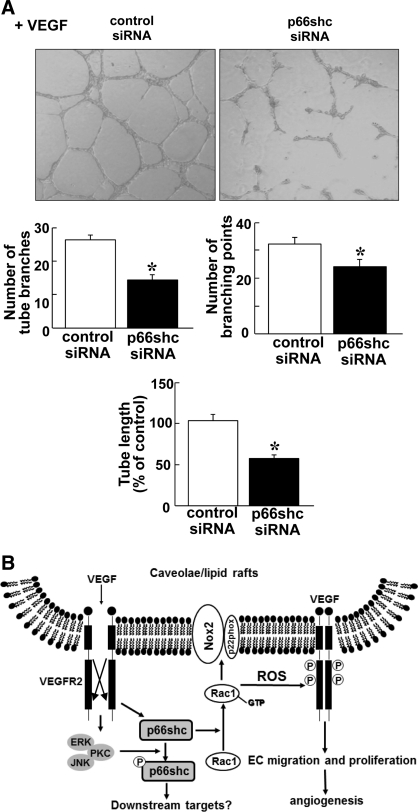
p66Shc, a longevity adaptor protein, is demonstrated as a key regulator of reactive oxygen species (ROS) metabolism involved in aging and cardiovascular diseases. Vascular endothelial growth factor (VEGF) stimulates endothelial cell (EC) migration and proliferation primarily through the VEGF receptor-2 (VEGFR2). We have shown that ROS derived from Rac1-dependent NADPH oxidase are involved in VEGFR2 autophosphorylation and angiogenic-related responses in ECs. However, a role of p66Shc in VEGF signaling and physiological responses in ECs is unknown. Here we show that VEGF promotes p66Shc phosphorylation at Ser36 through the JNK/ERK or PKC pathway as well as Rac1 binding to a nonphosphorylated form of p66Shc in ECs. Depletion of endogenous p66Shc with short interfering RNA inhibits VEGF-induced Rac1 activity and ROS production. Fractionation of caveolin-enriched lipid raft demonstrates that p66Shc plays a critical role in VEGFR2 phosphorylation in caveolae/lipid rafts as well as downstream p38MAP kinase activation. This in turn stimulates VEGF-induced EC migration, proliferation, and capillary-like tube formation. These studies uncover a novel role of p66Shc as a positive regulator for ROS-dependent VEGFR2 signaling linked to angiogenesis in ECs and suggest p66Shc as a potential therapeutic target for various angiogenesis-dependent diseases.
Figures



References
-
- Arany I, Faisal A, Nagamine Y, Safirstein RL. p66Shc inhibits pro-survival epidermal growth factor receptor/ERK signaling during severe oxidative stress in mouse renal proximal tubule cells. J Biol Chem 283: 6110–6117, 2008 - PubMed
-
- Babior BM. The NADPH oxidase of endothelial cells. IUBMB Life 50: 267–269, 2000 - PubMed
-
- Camici GG, Schiavoni M, Francia P, Bachschmid M, Martin-Padura I, Hersberger M, Tanner FC, Pelicci P, Volpe M, Anversa P, Luscher TF, Cosentino F. Genetic deletion of p66(Shc) adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proc Natl Acad Sci USA 104: 5217–5222, 2007 - PMC - PubMed
-
- Carpi A, Menabo R, Kaludercic N, Pelicci P, Di Lisa F, Giorgio M. The cardioprotective effects elicited by p66(Shc) ablation demonstrate the crucial role of mitochondrial ROS formation in ischemia/reperfusion injury. Biochim Biophys Acta 1787: 774–780, 2009 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous