

Muscle oxygen transport and utilization in heart failure: implications for exercise (in)tolerance
- PMID: 22101528
- PMCID: PMC3311454
- DOI: 10.1152/ajpheart.00943.2011
Muscle oxygen transport and utilization in heart failure: implications for exercise (in)tolerance
Abstract
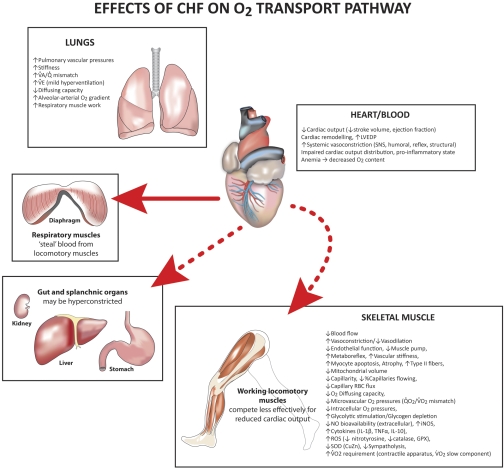
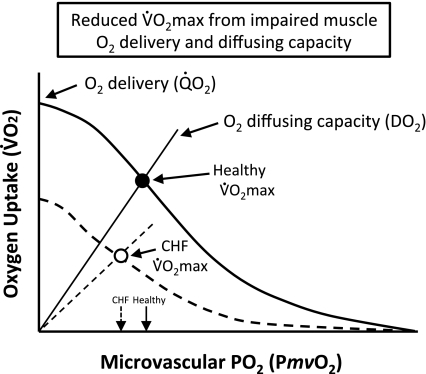
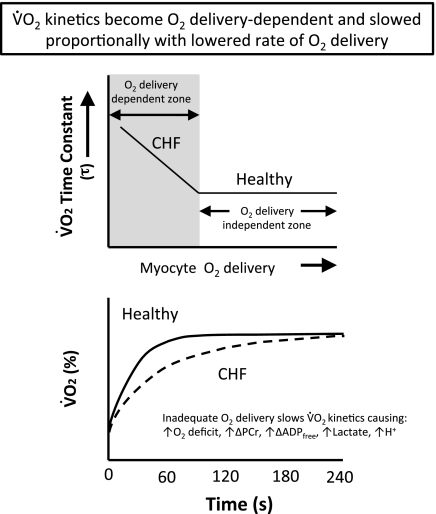
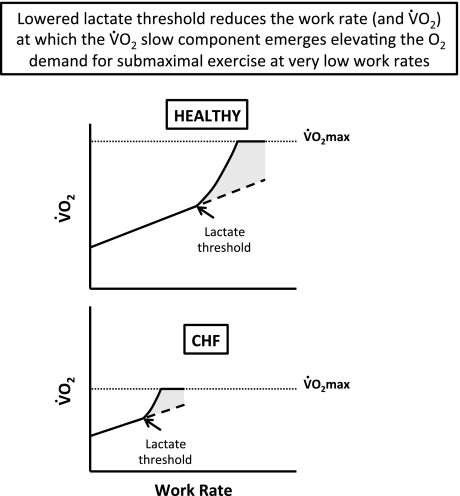
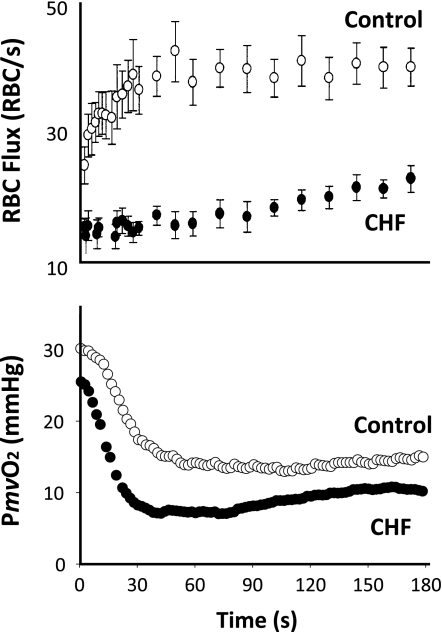
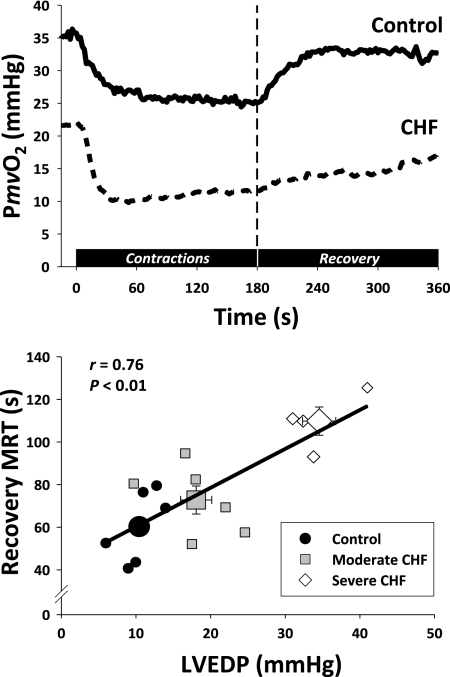
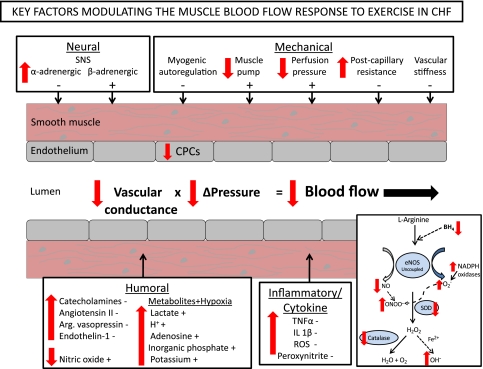
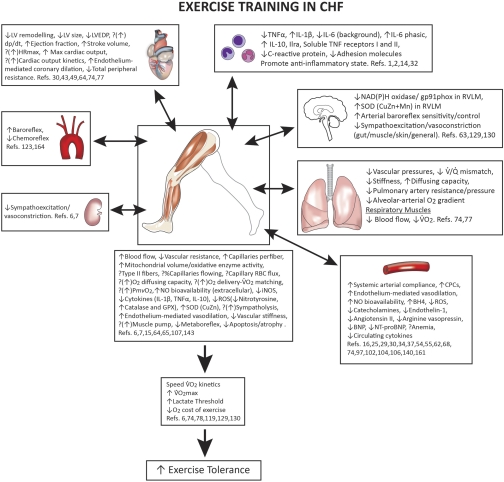
The defining characteristic of chronic heart failure (CHF) is an exercise intolerance that is inextricably linked to structural and functional aberrations in the O(2) transport pathway. CHF reduces muscle O(2) supply while simultaneously increasing O(2) demands. CHF severity varies from moderate to severe and is assessed commonly in terms of the maximum O(2) uptake, which relates closely to patient morbidity and mortality in CHF and forms the basis for Weber and colleagues' (167) classifications of heart failure, speed of the O(2) uptake kinetics following exercise onset and during recovery, and the capacity to perform submaximal exercise. As the heart fails, cardiovascular regulation shifts from controlling cardiac output as a means for supplying the oxidative energetic needs of exercising skeletal muscle and other organs to preventing catastrophic swings in blood pressure. This shift is mediated by a complex array of events that include altered reflex and humoral control of the circulation, required to prevent the skeletal muscle "sleeping giant" from outstripping the pathologically limited cardiac output and secondarily impacts lung (and respiratory muscle), vascular, and locomotory muscle function. Recently, interest has also focused on the dysregulation of inflammatory mediators including tumor necrosis factor-α and interleukin-1β as well as reactive oxygen species as mediators of systemic and muscle dysfunction. This brief review focuses on skeletal muscle to address the mechanistic bases for the reduced maximum O(2) uptake, slowed O(2) uptake kinetics, and exercise intolerance in CHF. Experimental evidence in humans and animal models of CHF unveils the microvascular cause(s) and consequences of the O(2) supply (decreased)/O(2) demand (increased) imbalance emblematic of CHF. Therapeutic strategies to improve muscle microvascular and oxidative function (e.g., exercise training and anti-inflammatory, antioxidant strategies, in particular) and hence patient exercise tolerance and quality of life are presented within their appropriate context of the O(2) transport pathway.
Figures
References
-
- Adamopoulos S, Parissis J, Karatzas D, Kroupis C, Georgiadis M, Karavolias G, Paraskevaidis J, Koniavitou K, Coats AJ, Kremastinos DT. Physical training modulates proinflammatory cytokines and the soluble Fas/soluble Fas ligand system in patients with chronic heart failure. J Am Coll Cardiol 39: 653–663, 2002 - PubMed
-
- Adamopoulos S, Parissis J, Kroupis C, Georgiadis M, Karatzas D, Karavolias G, Koniavitou K, Coats AJ, Kremastinos DT. Physical training reduces peripheral markers of inflammation in patients with chronic heart failure. Eur Heart J 22: 791–797, 2001 - PubMed
-
- Adams V, Jiang H, Yu J, Möbius-Winkler S, Fiehn E, Linke A, Weigl C, Schuler G, Hambrecht R. Apoptosis in skeletal myocytes of patients with chronic heart failure is associated with exercise intolerance. J Am Coll Cardiol 33: 959–965, 1999 - PubMed
-
- Adams V, Nehrhoff B, Späte U, Linke A, Schulze PC, Baur A, Gielen S, Hambrecht R, Schuler G. Induction of iNOS expression in skeletal muscle by IL-1beta and NFkappaB activation: an in vitro and in vivo study. Cardiovasc Res 54: 95–104, 2002 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials