

GenomeView: a next-generation genome browser
- PMID: 22102585
- PMCID: PMC3258165
- DOI: 10.1093/nar/gkr995
GenomeView: a next-generation genome browser
Abstract
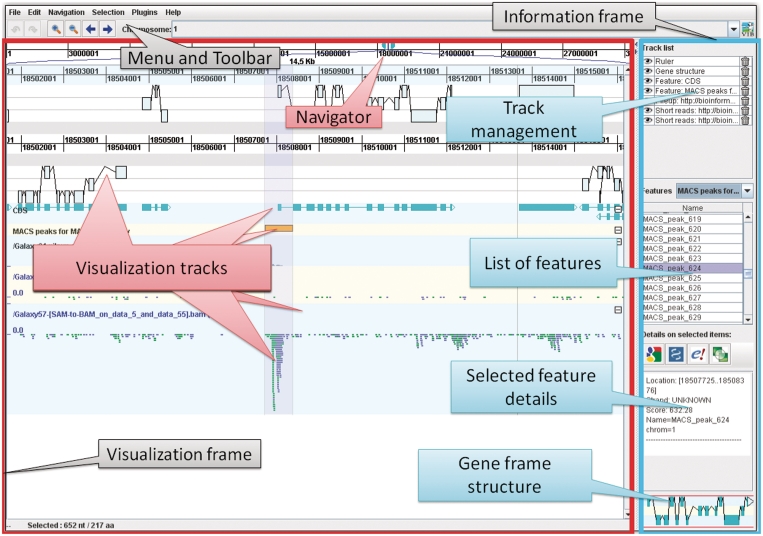
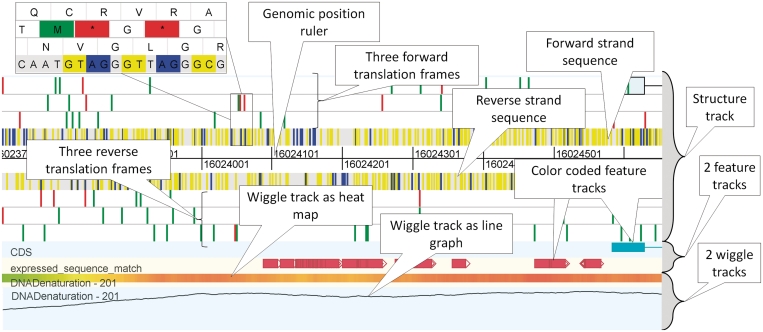
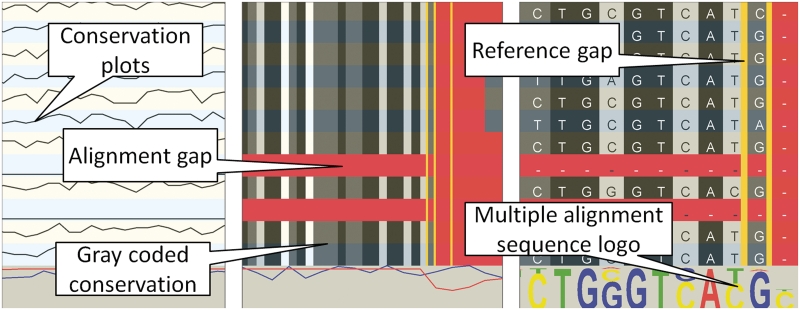
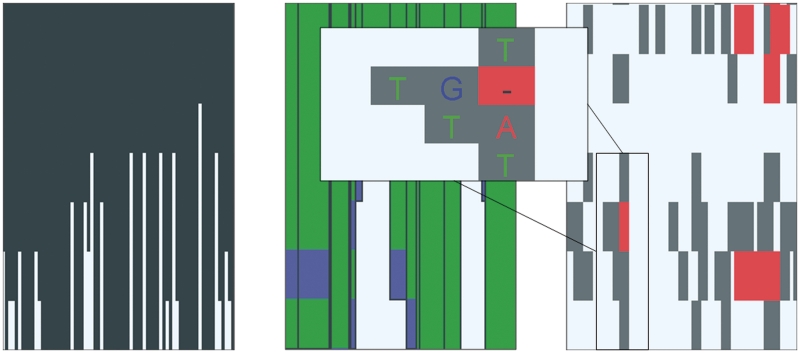
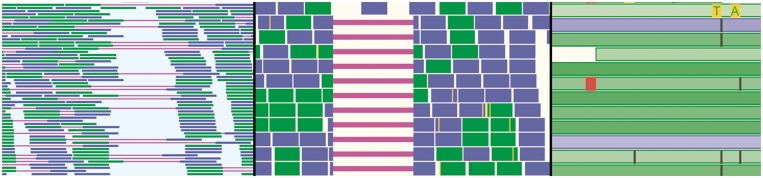
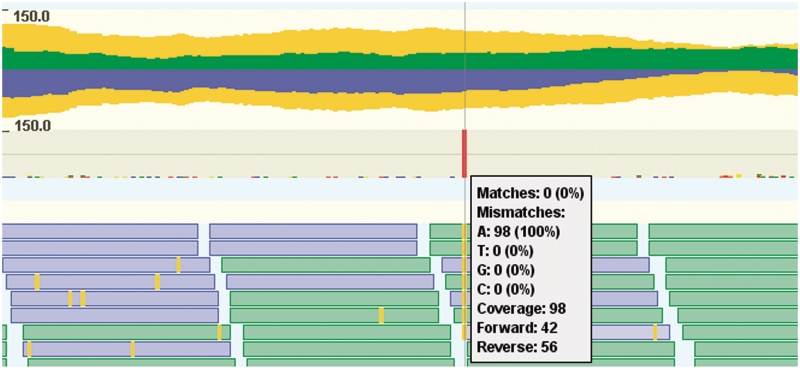
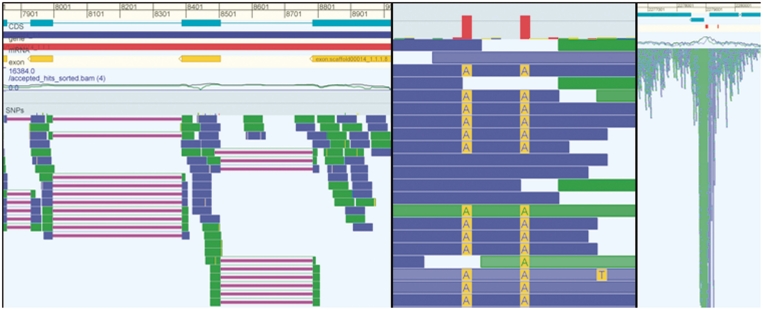
Due to ongoing advances in sequencing technologies, billions of nucleotide sequences are now produced on a daily basis. A major challenge is to visualize these data for further downstream analysis. To this end, we present GenomeView, a stand-alone genome browser specifically designed to visualize and manipulate a multitude of genomics data. GenomeView enables users to dynamically browse high volumes of aligned short-read data, with dynamic navigation and semantic zooming, from the whole genome level to the single nucleotide. At the same time, the tool enables visualization of whole genome alignments of dozens of genomes relative to a reference sequence. GenomeView is unique in its capability to interactively handle huge data sets consisting of tens of aligned genomes, thousands of annotation features and millions of mapped short reads both as viewer and editor. GenomeView is freely available as an open source software package.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
