

An RNA-Seq strategy to detect the complete coding and non-coding transcriptome including full-length imprinted macro ncRNAs
- PMID: 22102886
- PMCID: PMC3213133
- DOI: 10.1371/journal.pone.0027288
An RNA-Seq strategy to detect the complete coding and non-coding transcriptome including full-length imprinted macro ncRNAs
Abstract
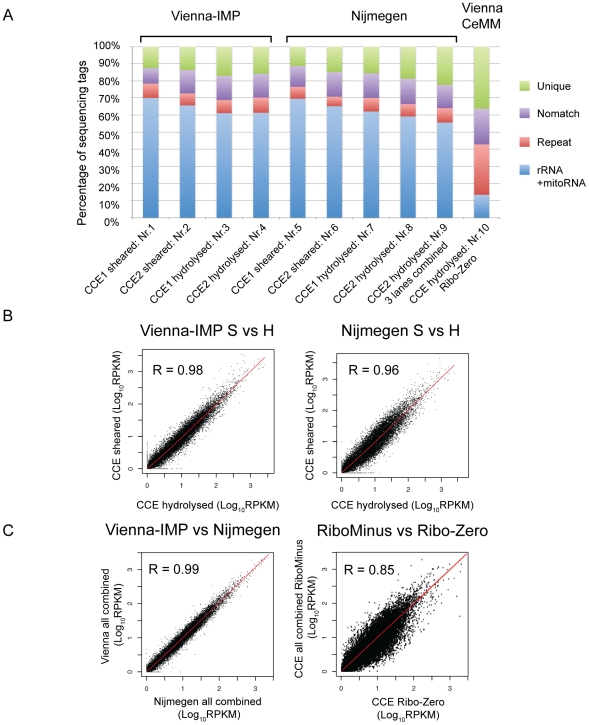
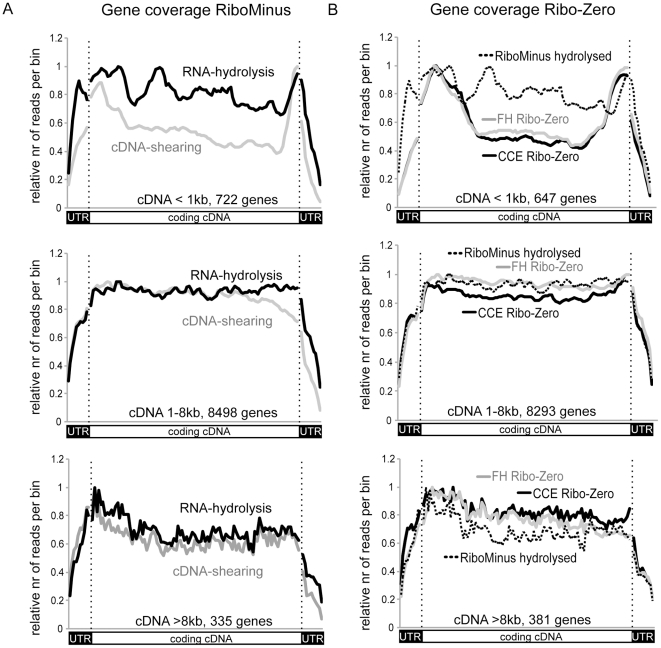
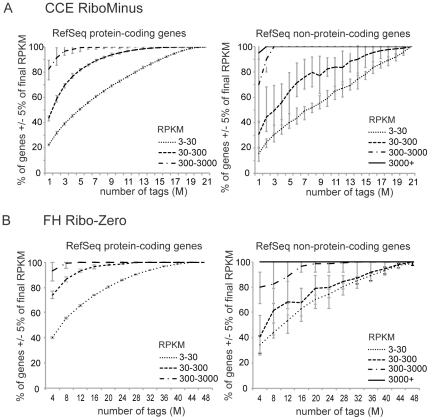
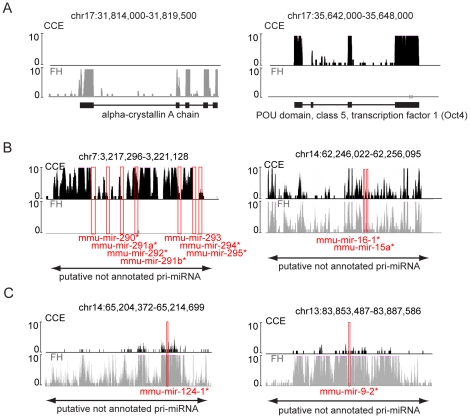
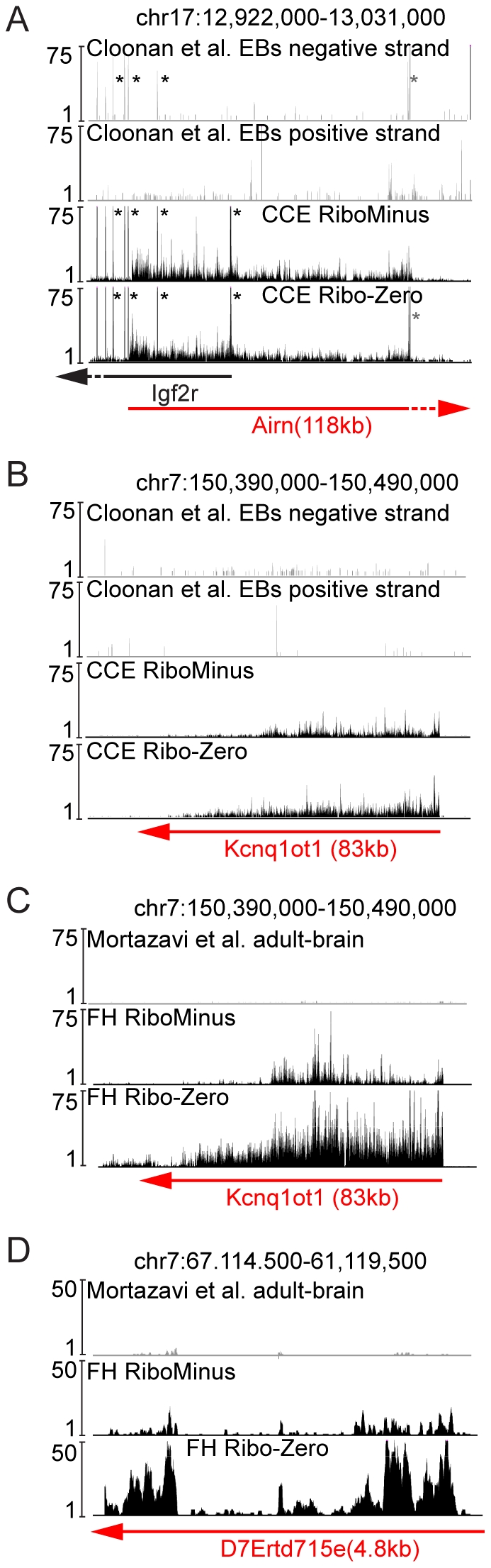
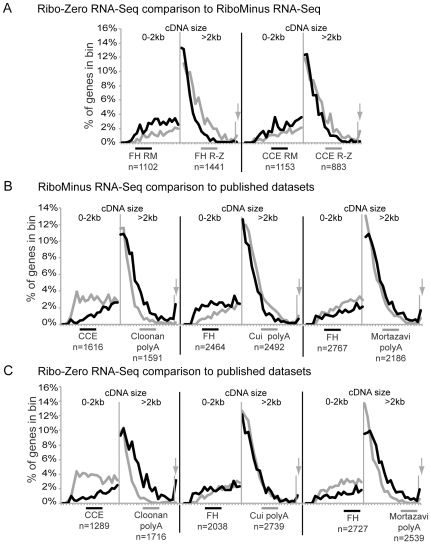
Imprinted macro non-protein-coding (nc) RNAs are cis-repressor transcripts that silence multiple genes in at least three imprinted gene clusters in the mouse genome. Similar macro or long ncRNAs are abundant in the mammalian genome. Here we present the full coding and non-coding transcriptome of two mouse tissues: differentiated ES cells and fetal head using an optimized RNA-Seq strategy. The data produced is highly reproducible in different sequencing locations and is able to detect the full length of imprinted macro ncRNAs such as Airn and Kcnq1ot1, whose length ranges between 80-118 kb. Transcripts show a more uniform read coverage when RNA is fragmented with RNA hydrolysis compared with cDNA fragmentation by shearing. Irrespective of the fragmentation method, all coding and non-coding transcripts longer than 8 kb show a gradual loss of sequencing tags towards the 3' end. Comparisons to published RNA-Seq datasets show that the strategy presented here is more efficient in detecting known functional imprinted macro ncRNAs and also indicate that standardization of RNA preparation protocols would increase the comparability of the transcriptome between different RNA-Seq datasets.
Conflict of interest statement
Figures
References
-
- Carninci P, Kasukawa T, Katayama S, Gough J, Frith MC, et al. The transcriptional landscape of the mammalian genome. Science. 2005;309:1559–1563. - PubMed
-
- Kapranov P, Cawley SE, Drenkow J, Bekiranov S, Strausberg RL, et al. Large-scale transcriptional activity in chromosomes 21 and 22. Science. 2002;296:916–919. - PubMed
-
- Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, et al. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. - PubMed
-
- Mattick JS, Taft RJ, Faulkner GJ. A global view of genomic information–moving beyond the gene and the master regulator. Trends in genetics : TIG. 2010;26:21–28. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
