

EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas
- PMID: 22105169
- PMCID: PMC3226010
- DOI: 10.1172/JCI60417
EGFR as a therapeutic target for human, canine, and mouse ACTH-secreting pituitary adenomas
Abstract
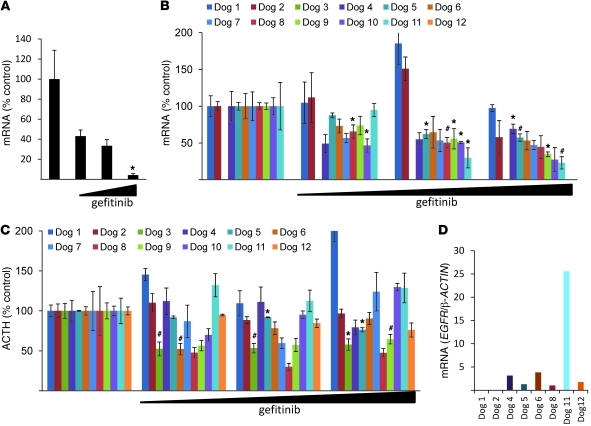
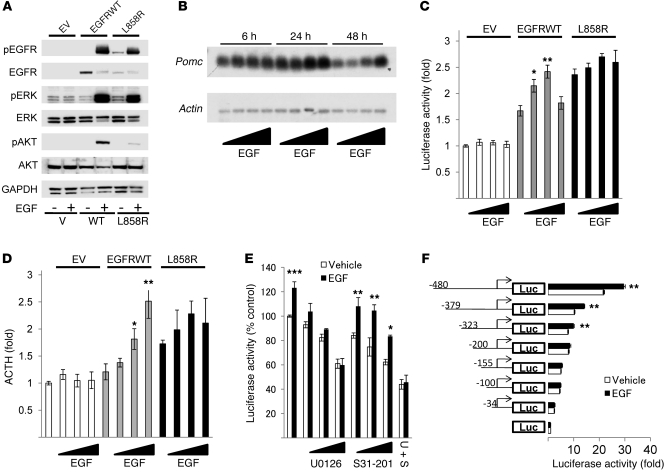
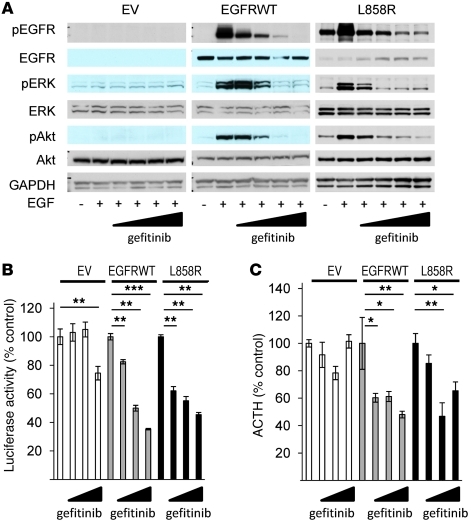
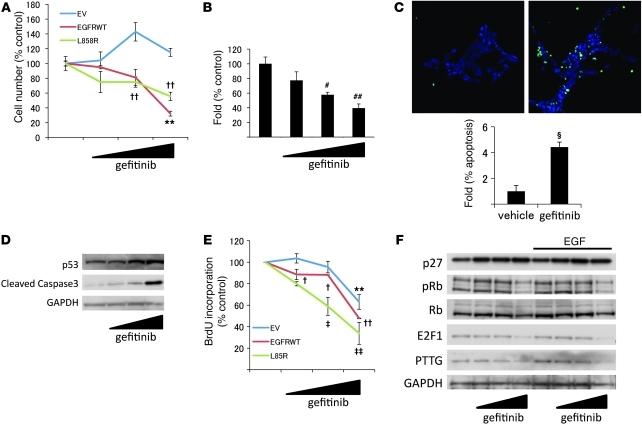
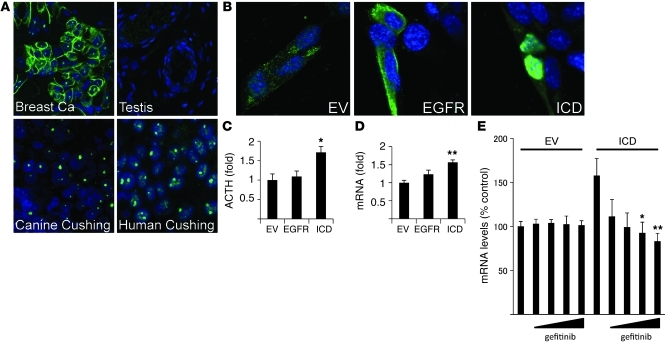
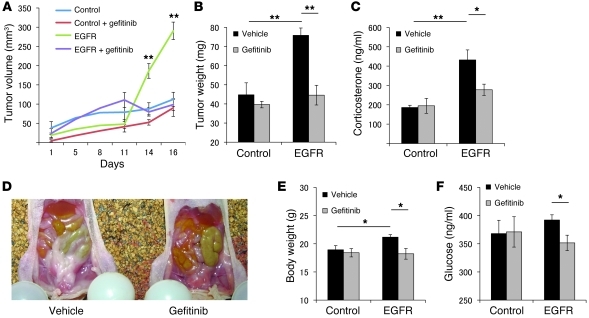
Cushing disease is a condition in which the pituitary gland releases excessive adrenocorticotropic hormone (ACTH) as a result of an adenoma arising from the ACTH-secreting cells in the anterior pituitary. ACTH-secreting pituitary adenomas lead to hypercortisolemia and cause significant morbidity and mortality. Pituitary-directed medications are mostly ineffective, and new treatment options are needed. As these tumors express EGFR, we tested whether EGFR might provide a therapeutic target for Cushing disease. Here, we show that in surgically resected human and canine corticotroph cultured tumors, blocking EGFR suppressed expression of proopiomelanocortin (POMC), the ACTH precursor. In mouse corticotroph EGFR transfectants, ACTH secretion was enhanced, and EGF increased Pomc promoter activity, an effect that was dependent on MAPK. Blocking EGFR activity with gefitinib, an EGFR tyrosine kinase inhibitor, attenuated Pomc expression, inhibited corticotroph tumor cell proliferation, and induced apoptosis. As predominantly nuclear EGFR expression was observed in canine and human corticotroph tumors, we preferentially targeted EGFR to mouse corticotroph cell nuclei, which resulted in higher Pomc expression and ACTH secretion, both of which were inhibited by gefitinib. In athymic nude mice, EGFR overexpression enhanced the growth of explanted ACTH-secreting tumors and further elevated serum corticosterone levels. Gefitinib treatment decreased both tumor size and corticosterone levels; it also reversed signs of hypercortisolemia, including elevated glucose levels and excess omental fat. These results indicate that inhibiting EGFR signaling may be a novel strategy for treating Cushing disease.
Figures
Comment in
-
A new medical therapy for Cushing disease?J Clin Invest. 2011 Dec;121(12):4621-3. doi: 10.1172/JCI61127. Epub 2011 Nov 21. J Clin Invest. 2011. PMID: 22105164 Free PMC article.
References
-
- Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet. 2006;367(9522):1605–1617. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
