

Progress and problems in muscle glycogenoses
- PMID: 22106711
- PMCID: PMC3235878
Progress and problems in muscle glycogenoses
Abstract
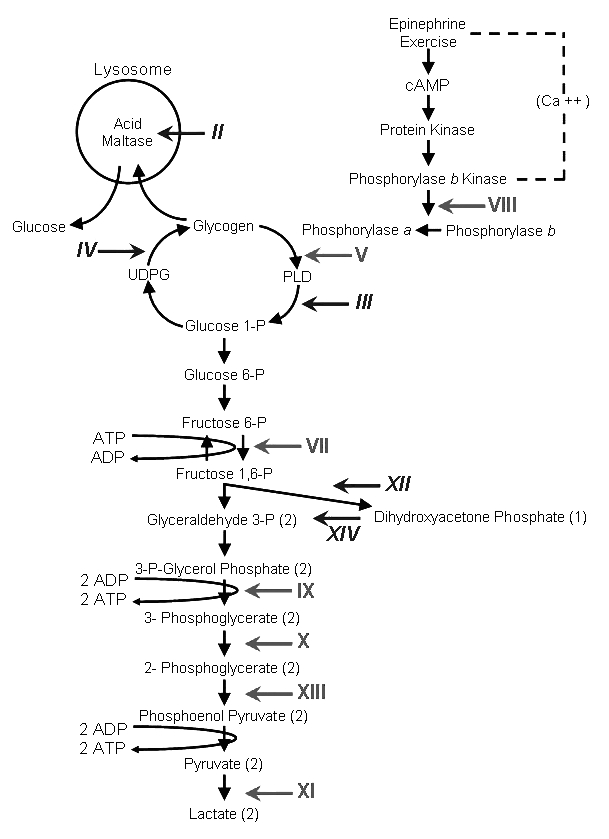
In this selective review, we consider a number of unsolved questions regarding the glycogen storage diseases (GSD). Thus, the pathogenesis of Pompe disease (GSD II) is not simply explained by excessive intralysosomal glycogen storage and may relate to a more general dysfunction of autophagy. It is not clear why debrancher deficiency (GSD III) causes fixed myopathy rather than exercise intolerance, unless this is due to the frequent accompanying neuropathy. The infantile neuromuscular presentation of branching enzyme deficiency (GSD IV) is underdiagnosed and is finally getting the attention it deserves. On the other hand, the late-onset variant of GSD IV (adult polyglucosan body disease APBD) is one of several polyglucosan disorders (including Lafora disease) due to different etiologies. We still do not understand the clinical heterogeneity of McArdle disease (GSD V) or the molecular basis of the rare fatal infantile form. Similarly, the multisystemic infantile presentation of phosphofructokinase deficiency (GSD VII) is a conundrum. We observed an interesting association between phosphoglycerate kinase deficiency (GSD IX) and juvenile Parkinsonism, which is probably causal rather than casual. Also unexplained is the frequent and apparently specific association of phosphoglycerate mutase deficiency (GSD X) and tubular aggregates. By paying more attention to problems than to progress, we aimed to look to the future rather than to the past.
Figures
References
-
- Favini Sacerdoti F, DiMauro S, Angelini Dobrilla C. Un caso di glicogenosi tipo II (m. di Pompe). Studio clinico, morfologico e biochimico. Acta Paediatr Latina. 1967;20:651–677.
-
- DiMauro S, Rowland LP, DiMauro PM. Control of glycogen metabolism in human muscle. Arch Neurol. 1970;23:534–540. - PubMed
-
- Askanas V, Engel WK, DiMauro S, et al. Adult-onset acid maltase deficiency. Morphological and biochemical abnormalities reproduced in cultured muscle. New Engl J Med. 1976;294:573–578. - PubMed
-
- Spiegel R, Area Gomez E, Akman HO, et al. Myopathic form of phosphoglycerate kinase (PGK) deficiency: A new case and pathogenic considerations. Neuromusc Disord. 2009;19:207–211. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous