

Optimal deconvolution of transcriptional profiling data using quadratic programming with application to complex clinical blood samples
- PMID: 22110609
- PMCID: PMC3217948
- DOI: 10.1371/journal.pone.0027156
Optimal deconvolution of transcriptional profiling data using quadratic programming with application to complex clinical blood samples
Abstract
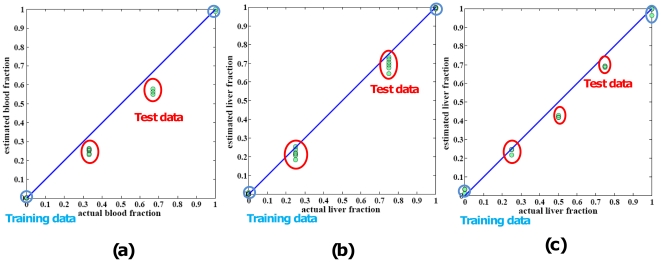
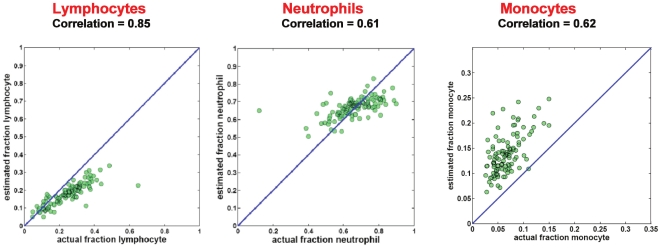
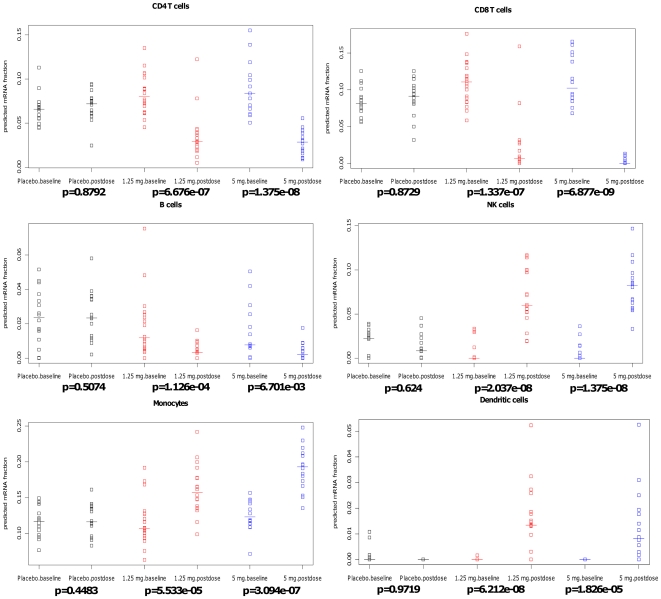
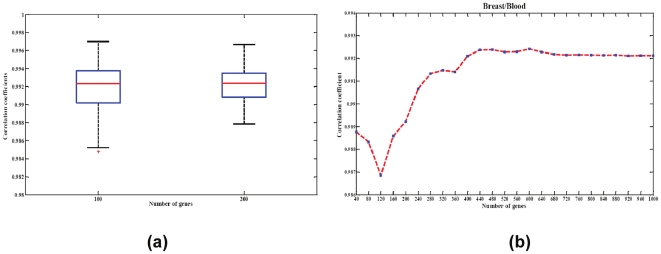
Large-scale molecular profiling technologies have assisted the identification of disease biomarkers and facilitated the basic understanding of cellular processes. However, samples collected from human subjects in clinical trials possess a level of complexity, arising from multiple cell types, that can obfuscate the analysis of data derived from them. Failure to identify, quantify, and incorporate sources of heterogeneity into an analysis can have widespread and detrimental effects on subsequent statistical studies.We describe an approach that builds upon a linear latent variable model, in which expression levels from mixed cell populations are modeled as the weighted average of expression from different cell types. We solve these equations using quadratic programming, which efficiently identifies the globally optimal solution while preserving non-negativity of the fraction of the cells. We applied our method to various existing platforms to estimate proportions of different pure cell or tissue types and gene expression profilings of distinct phenotypes, with a focus on complex samples collected in clinical trials. We tested our methods on several well controlled benchmark data sets with known mixing fractions of pure cell or tissue types and mRNA expression profiling data from samples collected in a clinical trial. Accurate agreement between predicted and actual mixing fractions was observed. In addition, our method was able to predict mixing fractions for more than ten species of circulating cells and to provide accurate estimates for relatively rare cell types (<10% total population). Furthermore, accurate changes in leukocyte trafficking associated with Fingolomid (FTY720) treatment were identified that were consistent with previous results generated by both cell counts and flow cytometry. These data suggest that our method can solve one of the open questions regarding the analysis of complex transcriptional data: namely, how to identify the optimal mixing fractions in a given experiment.
Conflict of interest statement
Figures
References
-
- Liotta L, Petricoin E. Molecular profiling of human cancer. Nat Rev Genet. 2000;1:48–56. - PubMed
-
- Coleman WB, Tsongalis GJ. 2009. Molecular Pathology: The Molecular Basis of Human Disease: Academic Press; 1 edition (March 16, 2009)
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
