

A large family with Carney complex caused by the S147G PRKAR1A mutation shows a unique spectrum of disease including adrenocortical cancer
- PMID: 22112814
- PMCID: PMC3275364
- DOI: 10.1210/jc.2011-2244
A large family with Carney complex caused by the S147G PRKAR1A mutation shows a unique spectrum of disease including adrenocortical cancer
Abstract
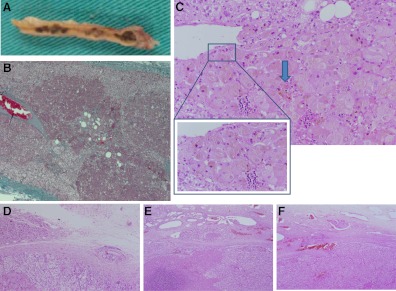
Context: Most tumors in Carney complex (CNC) are benign, including primary pigmented nodular adrenocortical disease (PPNAD), the main endocrine tumor in CNC. Adrenocortical cancer (AC) has never been observed in the syndrome. Herein, we describe a large Azorean family with CNC caused by a point mutation in the PRKAR1A gene coding for type 1-α (RIα) regulatory subunit of the cAMP-dependent protein kinase A, in which the index patient presented with AC.
Objective: We studied the genotype-phenotype correlation in CNC.
Design and setting: We reported on case series and in vitro testing of the PRKAR1A mutation in a tertiary care referral center.
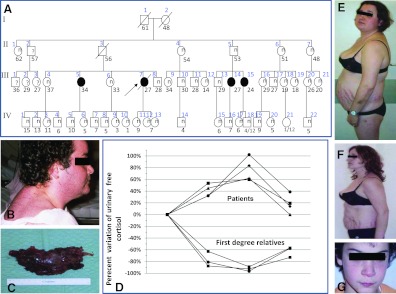
Patients: Twenty-two members of a family were investigated for Cushing syndrome and other CNC components; their DNA was sequenced for PRKAR1A mutations.
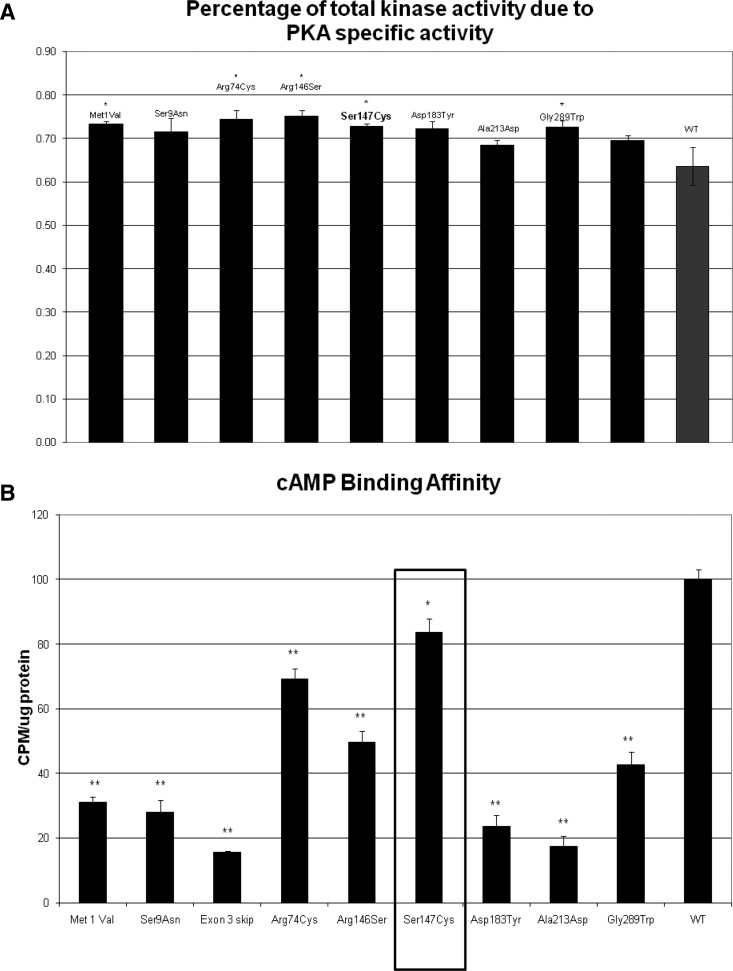
Results: Cushing syndrome due to PPNAD occurred in four patients, including the proposita who presented with AC and three who had Cushing syndrome and/or PPNAD. Lentigines were found in six additional patients who did not have PPNAD. A base substitution (c.439A>G/p.S147G) in PRKAR1A was identified in the proposita, in the three others with PPNAD, in the proposita's twin daughters who had lentigines but no evidence of hypercortisolism, and in five other family members, including one without lentigines or evidence of hypercortisolism. Unlike in other RIα defects, loss of heterozygosity was not observed in AC. The S147G mutation was compared to other expressed PRKAR1A mutations; it led to decreased cAMP and catalytic subunit binding by RIα and increased protein kinase A activity in vitro.
Conclusions: In a large family with CNC, one amino acid substitution caused a spectrum of adrenal disease that ranged from lack of manifestations to cancer. PPNAD and AC were the only manifestations of CNC in these patients, in addition to lentigines. These data have implications for counseling patients with CNC and are significant in documenting the first case of AC in the context of PPNAD.
Figures
Comment in
-
Adrenocortical cancer in Carney complex: a paradigm of endocrine tumor progression or an association of genetic predisposing factors?J Clin Endocrinol Metab. 2012 Feb;97(2):387-90. doi: 10.1210/jc.2011-3327. J Clin Endocrinol Metab. 2012. PMID: 22312093 No abstract available.
References
-
- Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL. 1985. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore) 64:270–283 - PubMed
-
- Stratakis CA, Kirschner LS, Carney JA. 2001. Clinical and molecular features of Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab 86:4041–4046 - PubMed
-
- Kirschner LS, Sandrini F, Monbo J, Lin JP, Carney JA, Stratakis CA. 2000. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with Carney complex. Hum Mol Genet 9:3037–3046 - PubMed
-
- Stratakis CA, Sarlis N, Kirschner LS, Carney JA, Doppman JL, Nieman LK, Chrousos GP, Papanicolaou DA. 1999. Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease. Ann Intern Med 131:585–591 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
