

doi: 10.1073/pnas.1115123108.
Epub 2011 Nov 23.
A fast doubly hybrid density functional method close to chemical accuracy using a local opposite spin ansatz
Affiliations
- PMID: 22114187
- PMCID: PMC3250148
- DOI: 10.1073/pnas.1115123108
Item in Clipboard
A fast doubly hybrid density functional method close to chemical accuracy using a local opposite spin ansatz
Proc Natl Acad Sci U S A.
.
Abstract
We develop and validate the XYGJ-OS functional, based on the adiabatic connection formalism and Görling-Levy perturbation theory to second order and using the opposite-spin (OS) ansatz combined with locality of electron correlation. XYGJ-OS with local implementation scales as N(3) with an overall accuracy of 1.28 kcal/mol for thermochemistry, bond dissociation energies, reaction barrier heights, and nonbonded interactions, comparable to that of 1.06 kcal/mol for the accurate coupled-cluster based G3 method (scales as N(7)) and much better than many popular density functional theory methods: B3LYP (4.98), PBE0 (4.36), and PBE (12.10).
Conflict of interest statement
The authors declare no conflict of interest.
Figures
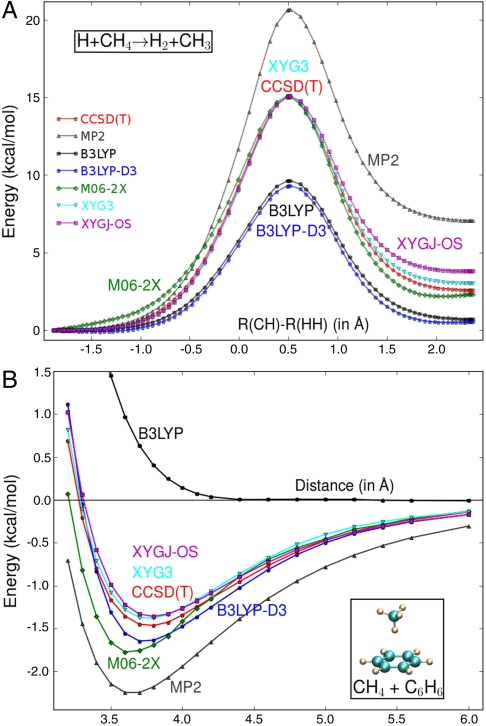
(A) Potential energy curves for H + CH4 → H2 + CH3 reaction along the reaction coordinate of [R(CH)-R(HH)] (in Å). (B) Intermolecular potentials for CH4-C6H6 complex. R is the distance from the carbon of CH4 to the ring center of C6H6.
References
-
- Curtiss LA, Redfern PC, Raghavachari K, Pople JA. Gaussian-3X (G3X) theory: use of improved geometries, zero-point energies, and Hartree-Fock basis sets. J Chem Phys. 2001;114:108–117.
-
- Curtiss LA, Redfern PC, Raghavachari K, Pople JA. Assessment of Gaussian-2 and density functional theories for the computation of ionization potentials and electron affinities. J Chem Phys. 1998;109:42–55.
-
- Yang WT. Direct calculation of electron density in density-functional theory. Phys Rev Lett. 1991;66:1438–1441. - PubMed
-
- Hohenberg P, Kohn W. Inhomogeneous electron gas. Phys Rev B. 1964;3:864–871.
-
- Kohn W, Sham LJ. Self-consistent equations including exchange and correlation effects. Phys Rev A. 1965;4:1133–1138.
Publication types
LinkOut - more resources
Full Text Sources
