

The loss of c-Jun N-terminal protein kinase activity prevents the amyloidogenic cleavage of amyloid precursor protein and the formation of amyloid plaques in vivo
- PMID: 22114267
- PMCID: PMC6623849
- DOI: 10.1523/JNEUROSCI.4491-11.2011
The loss of c-Jun N-terminal protein kinase activity prevents the amyloidogenic cleavage of amyloid precursor protein and the formation of amyloid plaques in vivo
Abstract
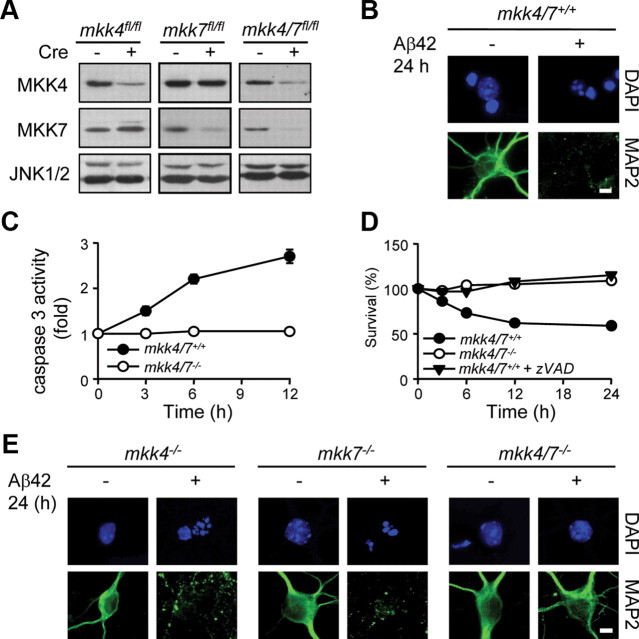
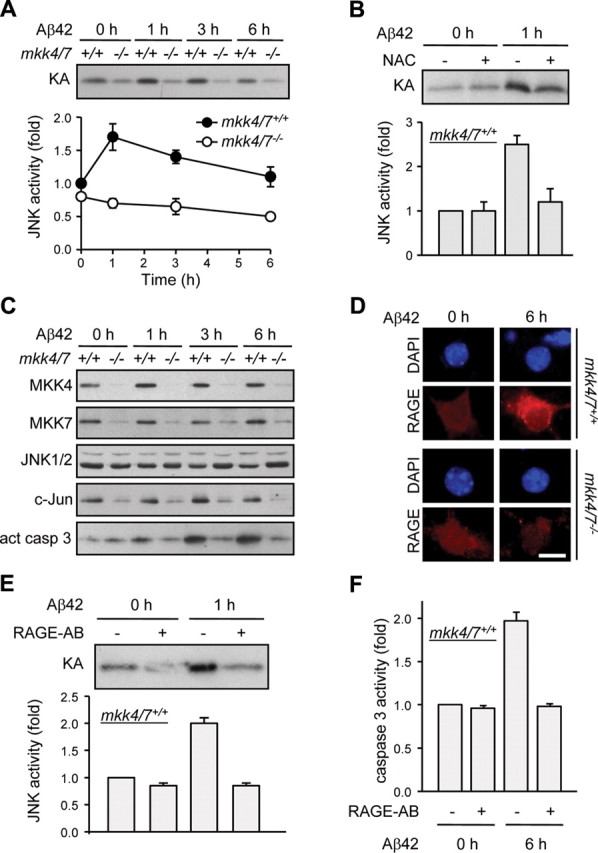
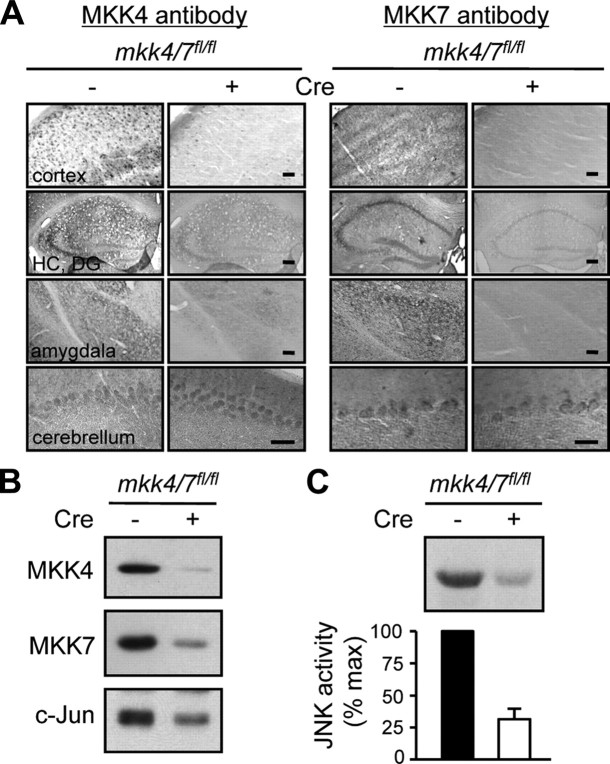
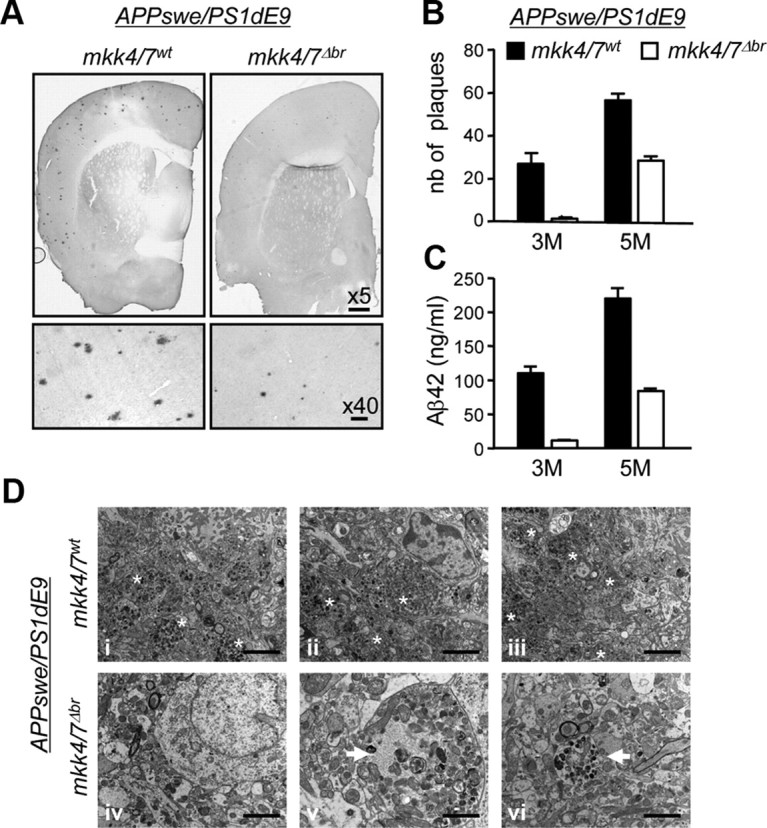
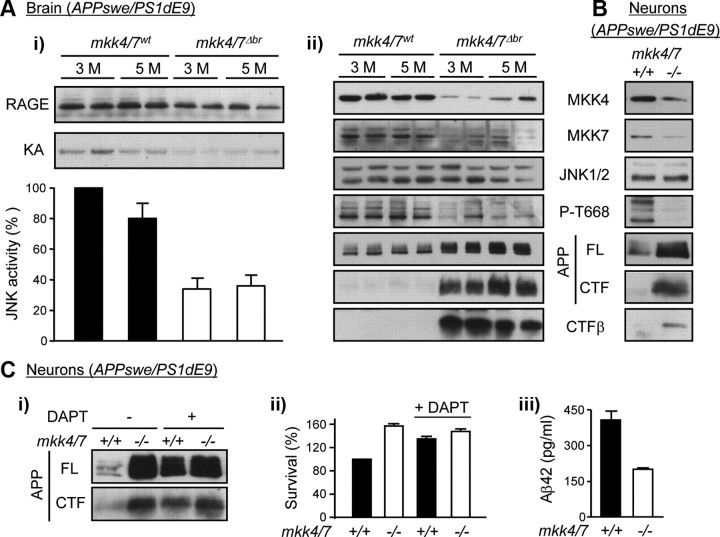
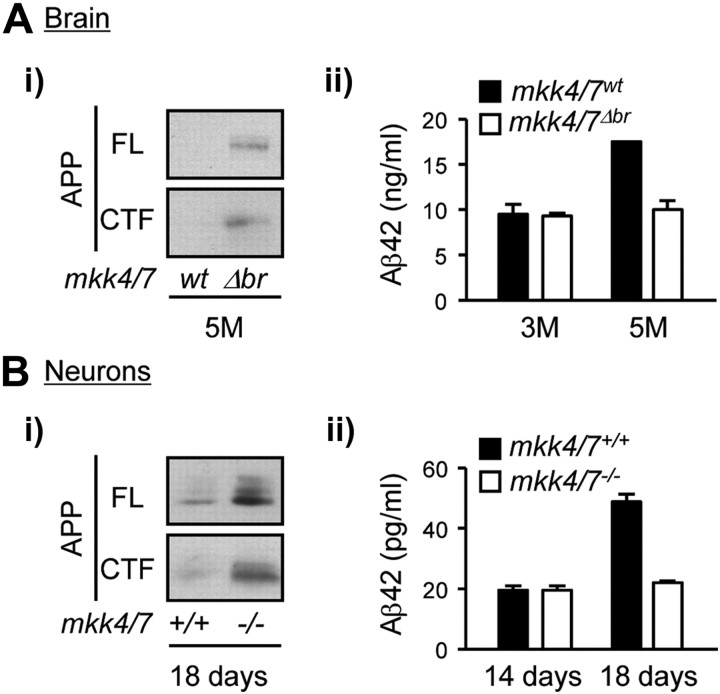
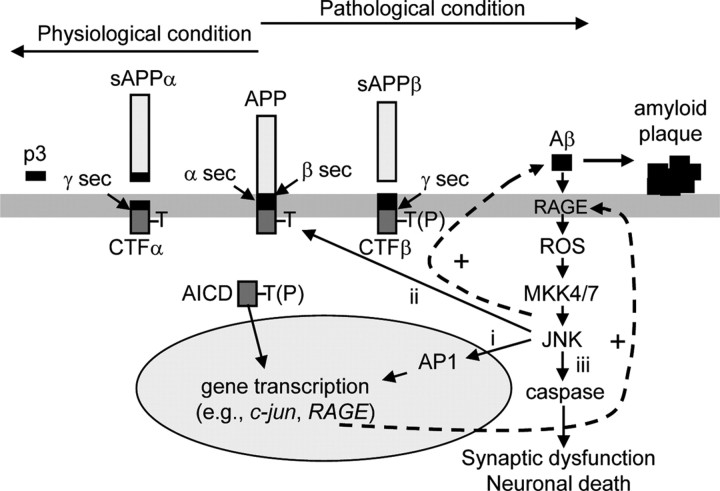
Phosphorylation plays a central role in the dynamic regulation of the processing of the amyloid precursor protein (APP) and the production of amyloid-β (Aβ), one of the clinically most important factors that determine the onset of Alzheimer's disease (AD). This has led to the hypothesis that aberrant Aβ production associated with AD results from regulatory defects in signal transduction. However, conflicting findings have raised a debate over the identity of the signaling pathway that controls APP metabolism. Here, we demonstrate that activation of the c-Jun N-terminal protein kinase (JNK) is essential for mediating the apoptotic response of neurons to Aβ. Furthermore, we discovered that the functional loss of JNK signaling in neurons significantly decreased the number of amyloid plaques present in the brain of mice carrying familial AD-linked mutant genes. This correlated with a reduction in Aβ production. Biochemical analyses indicate that the phosphorylation of APP at threonine 668 by JNK is required for γ-mediated cleavage of the C-terminal fragment of APP produced by β-secretase. Overall, this study provides genetic evidence that JNK signaling is required for the formation of amyloid plaques in vivo. Therefore, inhibition of increased JNK activity associated with aging or with a pathological condition constitutes a potential strategy for the treatment of AD.
Figures
References
-
- Akiyama H, Shin RW, Uchida C, Kitamoto T, Uchida T. Pin1 promotes production of Alzheimer's amyloid beta from beta-cleaved amyloid precursor protein. Biochem Biophys Res Commun. 2005;336:521–529. - PubMed
-
- Ando K, Iijima KI, Elliott JI, Kirino Y, Suzuki T. Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of beta-amyloid. J Biol Chem. 2001;276:40353–40361. - PubMed
-
- Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 1994;77:817–827. - PubMed
-
- Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68:270–281. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous