

Replication-competent influenza A virus that encodes a split-green fluorescent protein-tagged PB2 polymerase subunit allows live-cell imaging of the virus life cycle
- PMID: 22114331
- PMCID: PMC3264389
- DOI: 10.1128/JVI.05820-11
Replication-competent influenza A virus that encodes a split-green fluorescent protein-tagged PB2 polymerase subunit allows live-cell imaging of the virus life cycle
Abstract
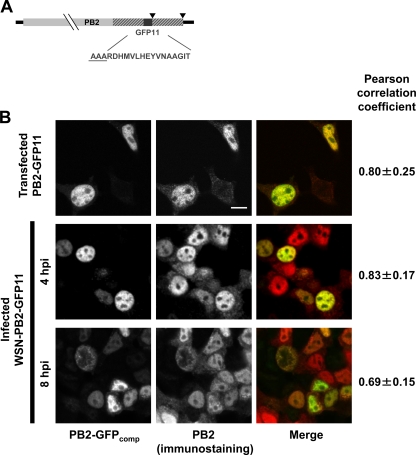
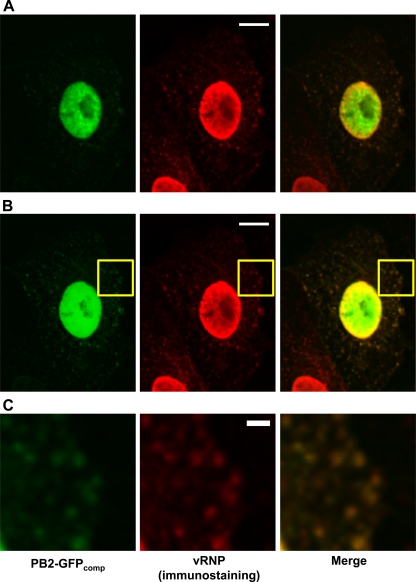
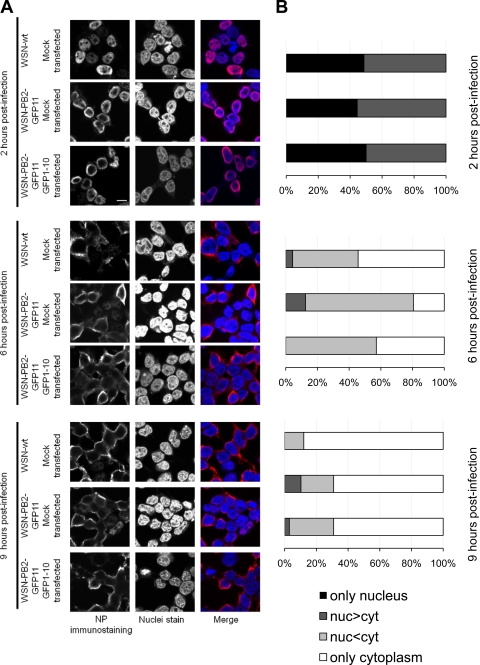
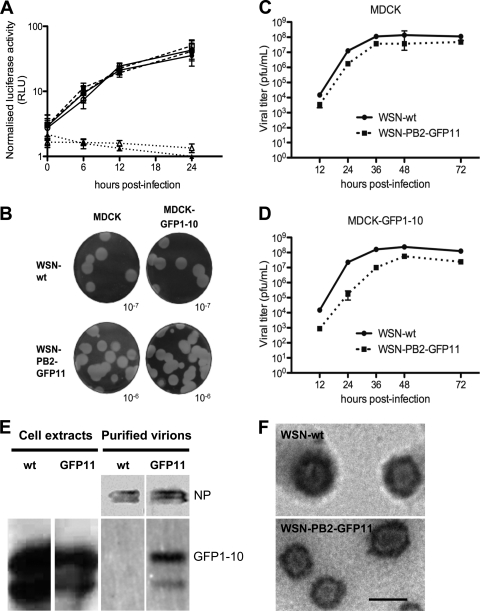
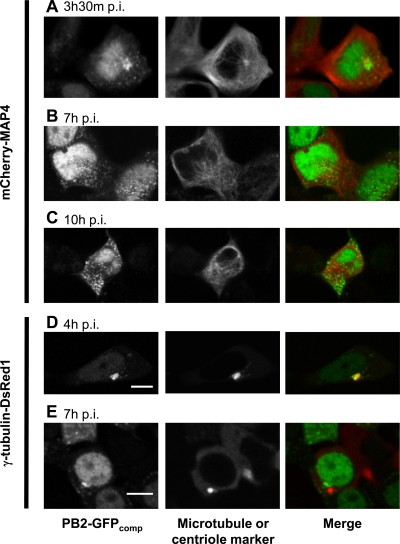
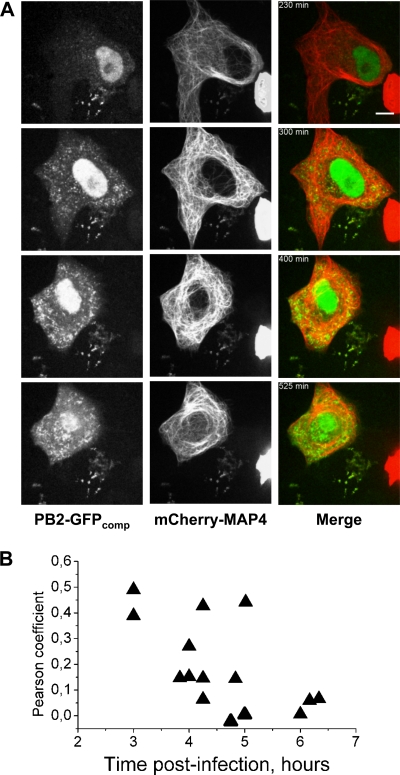
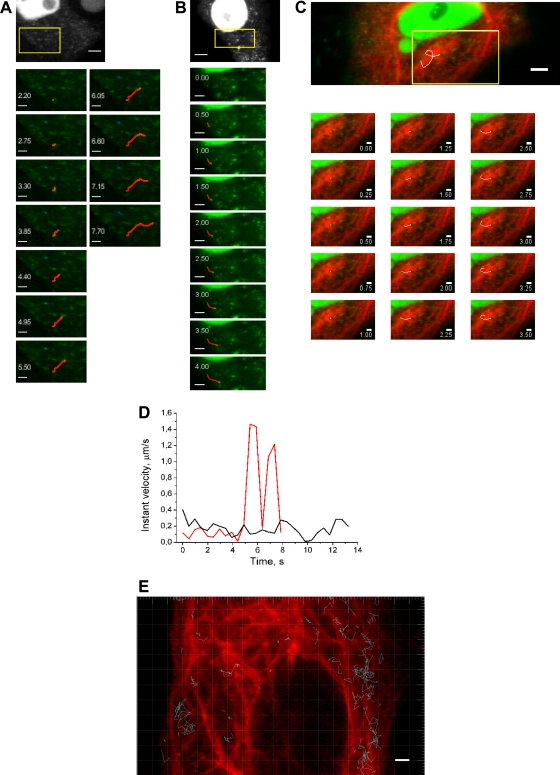
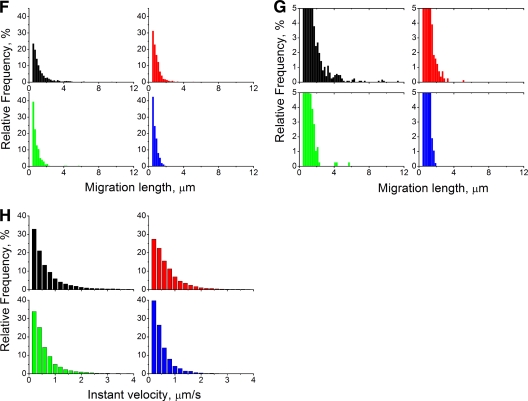
Studies on the intracellular trafficking of influenza virus ribonucleoproteins are currently limited by the lack of a method enabling their visualization during infection in single cells. This is largely due to the difficulty of encoding fluorescent fusion proteins within the viral genome. To circumvent this limitation, we used the split-green fluorescent protein (split-GFP) system (S. Cabantous, T. C. Terwilliger, and G. S. Waldo, Nat. Biotechnol. 23:102-107, 2005) to produce a quasi-wild-type recombinant A/WSN/33/influenza virus which allows expression of individually fluorescent PB2 polymerase subunits in infected cells. The viral PB2 proteins were fused to the 16 C-terminal amino acids of the GFP, whereas the large transcomplementing GFP fragment was supplied by transient or stable expression in cultured cells that were permissive to infection. This system was used to characterize the intranuclear dynamics of PB2 by fluorescence correlation spectroscopy and to visualize the trafficking of viral ribonucleoproteins (vRNPs) by dynamic light microscopy in live infected cells. Following nuclear export, vRNPs showed a transient pericentriolar accumulation and intermittent rapid (∼1 μm/s), directional movements in the cytoplasm, dependent on both microtubules and actin filaments. Our data establish the potential of split-GFP-based recombinant viruses for the tracking of viral proteins during a quasi-wild-type infection. This new virus, or adaptations of it, will be of use in elucidating many aspects of influenza virus host cell interactions as well as in screening for new antiviral compounds. Furthermore, the existence of cell lines stably expressing the complementing GFP fragment will facilitate applications to many other viral and nonviral systems.
Figures


References
-
- Cabantous S, Terwilliger TC, Waldo GS. 2005. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 23:102–107 - PubMed
-
- Crescenzo-Chaigne B, Naffakh N, van der Werf S. 1999. Comparative analysis of the ability of the polymerase complexes of influenza viruses type A, B and C to assemble into functional RNPs that allow expression and replication of heterotypic model RNA templates in vivo. Virology 265:342–353 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
