

Analysis of cellular localization and function of carboxy-terminal mutants of pendrin
- PMID: 22116356
- PMCID: PMC3709185
- DOI: 10.1159/000335105
Analysis of cellular localization and function of carboxy-terminal mutants of pendrin
Abstract
Background: Iodide uptake at the basolateral membrane and iodide efflux at the apical membrane of thyrocytes, essential steps in the biosynthesis of thyroid hormone, are stimulated by thyroid stimulating hormone (TSH). Pendrin (SLC26A4) is inserted into the apical membrane of thyrocytes and thought to be involved in mediating iodide efflux.
Methods: We determined the effects of carboxy-terminal mutations of pendrin on the cellular localization and the ability to transport iodide.
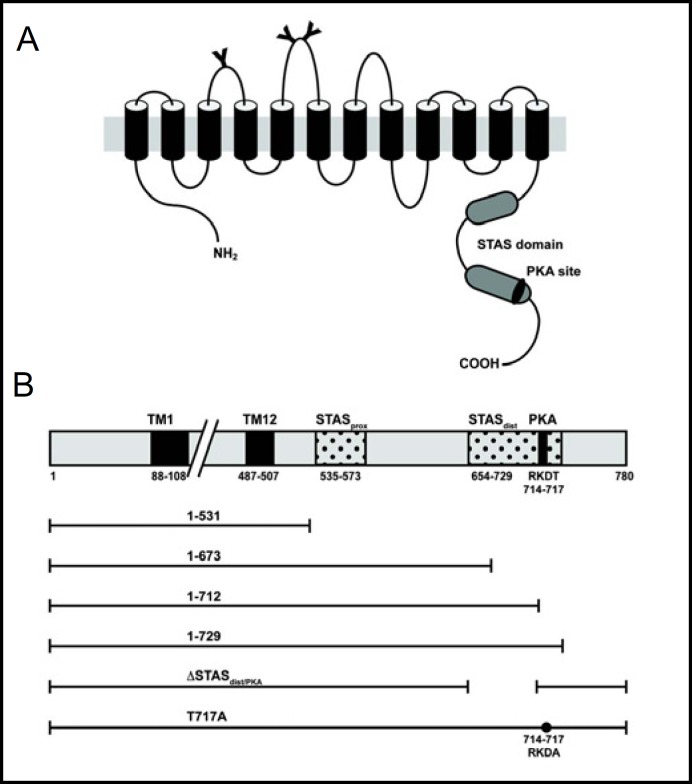
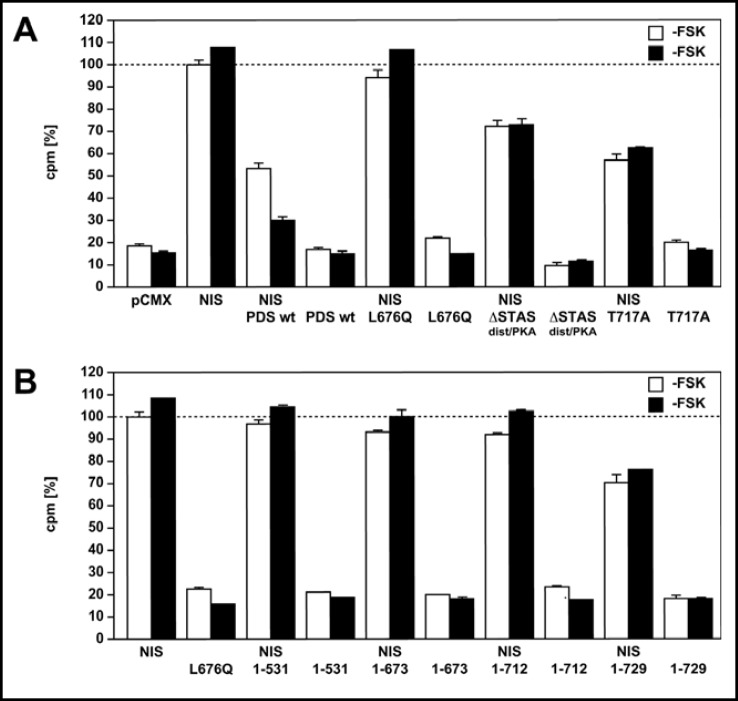
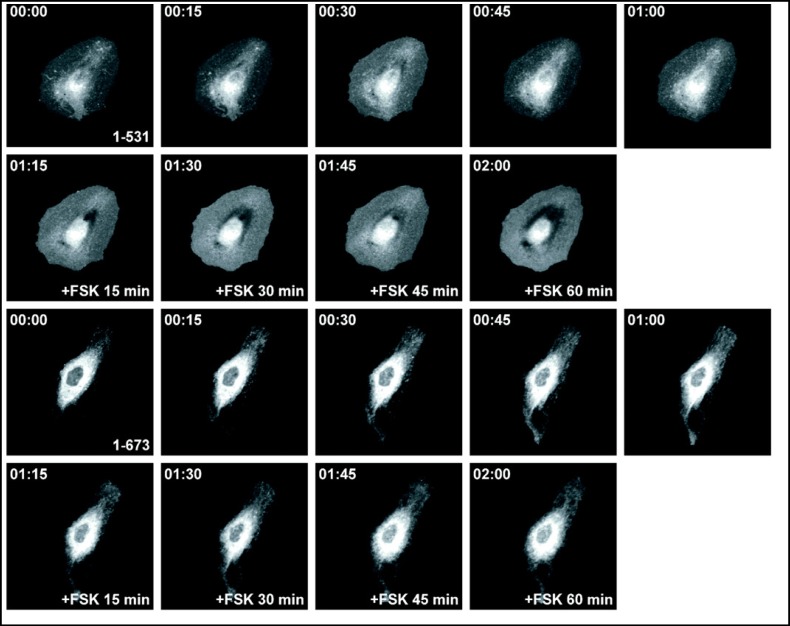
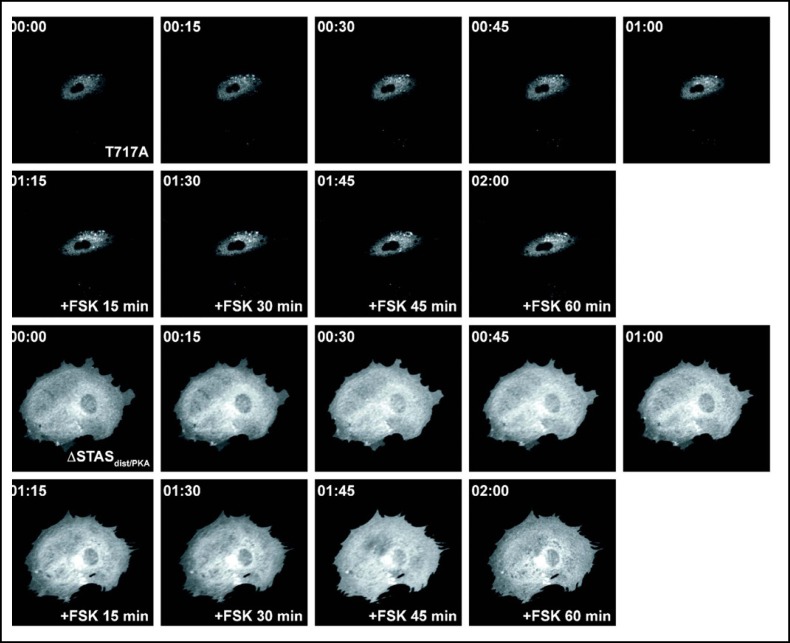
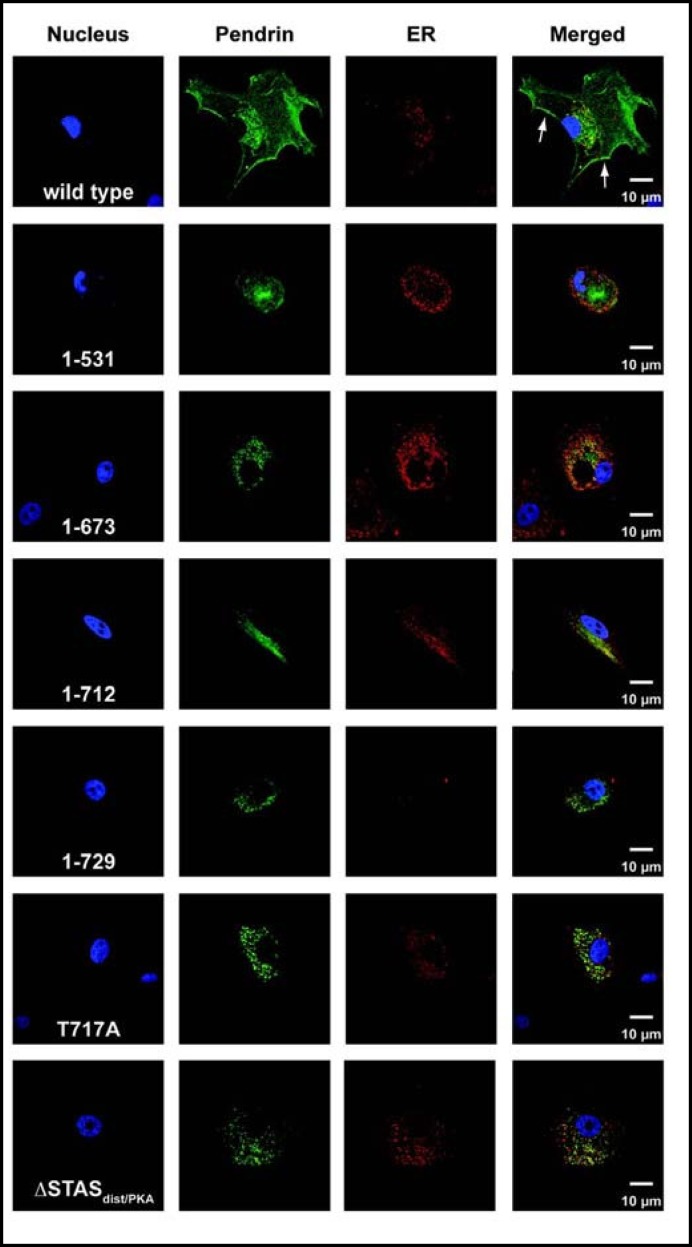
Results: After exposure to forskolin, the membrane abundance of wild type pendrin and iodide efflux increase. Truncation mutants lead to complete intracellular retention. Elimination of the distal part of the sulfate transporter and antisigma factor antagonist (STAS) domain with retention of the putative protein kinase A (PKA) phosphorylation site (RKDT 714-717) results in residual membrane insertion and a partial loss of function. Deletion of the PKA site results in decreased basal function and membrane insertion and abolishes the response to forskolin.
Conclusion: Pendrin membrane abundance and its ability to mediate iodide efflux increase after activation of the PKA pathway. Elimination of the PKA site abolishes the response to forskolin but partial basal function and membrane insertion are maintained.
Copyright © 2011 S. Karger AG, Basel.
Figures



Similar articles
-
TSH regulates pendrin membrane abundance and enhances iodide efflux in thyroid cells.Endocrinology. 2012 Jan;153(1):512-21. doi: 10.1210/en.2011-1548. Epub 2011 Nov 22. Endocrinology. 2012. PMID: 22109890 Free PMC article.
-
Controversies concerning the role of pendrin as an apical iodide transporter in thyroid follicular cells.Cell Physiol Biochem. 2011;28(3):485-90. doi: 10.1159/000335103. Epub 2011 Nov 18. Cell Physiol Biochem. 2011. PMID: 22116361 Review.
-
Functional characterization of pendrin in a polarized cell system. Evidence for pendrin-mediated apical iodide efflux.J Biol Chem. 2004 Mar 26;279(13):13004-10. doi: 10.1074/jbc.M313648200. Epub 2004 Jan 8. J Biol Chem. 2004. PMID: 14715652
-
Genetics and phenomics of Pendred syndrome.Mol Cell Endocrinol. 2010 Jun 30;322(1-2):83-90. doi: 10.1016/j.mce.2010.03.006. Epub 2010 Mar 15. Mol Cell Endocrinol. 2010. PMID: 20298745 Review.
-
Pendrin and anoctamin as mediators of apical iodide efflux in thyroid cells.Curr Opin Endocrinol Diabetes Obes. 2015 Oct;22(5):374-80. doi: 10.1097/MED.0000000000000188. Curr Opin Endocrinol Diabetes Obes. 2015. PMID: 26313899 Review.
Cited by
-
Comparative analysis of functional assay evidence use by ClinGen Variant Curation Expert Panels.Genome Med. 2019 Nov 29;11(1):77. doi: 10.1186/s13073-019-0683-1. Genome Med. 2019. PMID: 31783775 Free PMC article.
-
Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss.Hum Mutat. 2018 Nov;39(11):1593-1613. doi: 10.1002/humu.23630. Hum Mutat. 2018. PMID: 30311386 Free PMC article.
-
Protein phosphatase 1 coordinates CFTR-dependent airway epithelial HCO3- secretion by reciprocal regulation of apical and basolateral membrane Cl(-)-HCO3- exchangers.Br J Pharmacol. 2013 Apr;168(8):1946-60. doi: 10.1111/bph.12085. Br J Pharmacol. 2013. PMID: 23215877 Free PMC article.
-
A Novel Frameshift Mutation of SLC26A4 in a Korean Family With Nonsyndromic Hearing Loss and Enlarged Vestibular Aqueduct.Clin Exp Otorhinolaryngol. 2017 Mar;10(1):50-55. doi: 10.21053/ceo.2016.00430. Epub 2016 Jul 7. Clin Exp Otorhinolaryngol. 2017. PMID: 27384033 Free PMC article.
-
Atrophic thyroid follicles and inner ear defects reminiscent of cochlear hypothyroidism in Slc26a4-related deafness.Mamm Genome. 2014 Aug;25(7-8):304-16. doi: 10.1007/s00335-014-9515-1. Epub 2014 Apr 24. Mamm Genome. 2014. PMID: 24760582 Free PMC article.
References
-
- Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS) Nat Genet. 1997;17:411–422. - PubMed
-
- Bizhanova A, Kopp P. Genetics and phenomics of Pendred syndrome. Mol Cell Endocrinol. 2010;322:83–90. - PubMed
-
- Kopp P, Pesce L, Solis SJ. Pendred syndrome and iodide transport in the thyroid. Trends Endocrinol Metab. 2008;19:260–268. - PubMed
-
- Yoon JS, Park HJ, Yoo SY, Namkung W, Jo MJ, Koo SK, Park HY, Lee WS, Kim KH, Lee MG. Heterogeneity in the processing defect of SLC26A4 mutants. J Med Genet. 2008;45:411–419. - PubMed
-
- Rotman-Pikielny P, Hirschberg K, Maruvada P, Suzuki K, Royaux IE, Green ED, Kohn LD, Lippincott-Schwartz J, Yen PM. Retention of pendrin in the endoplasmic reticulum is a major mechanism for Pendred syndrome. Hum Mol Genet. 2002;11:2625–2633. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources