

p120-catenin modulates airway epithelial cell migration induced by cigarette smoke
- PMID: 22120634
- PMCID: PMC4066870
- DOI: 10.1016/j.bbrc.2011.11.048
p120-catenin modulates airway epithelial cell migration induced by cigarette smoke
Abstract
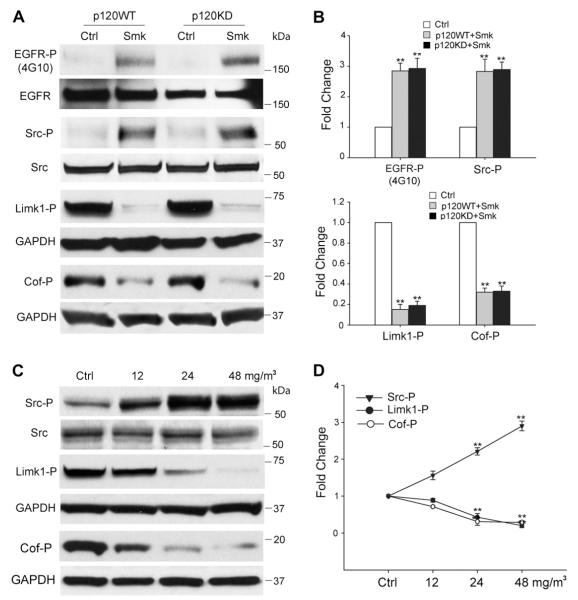
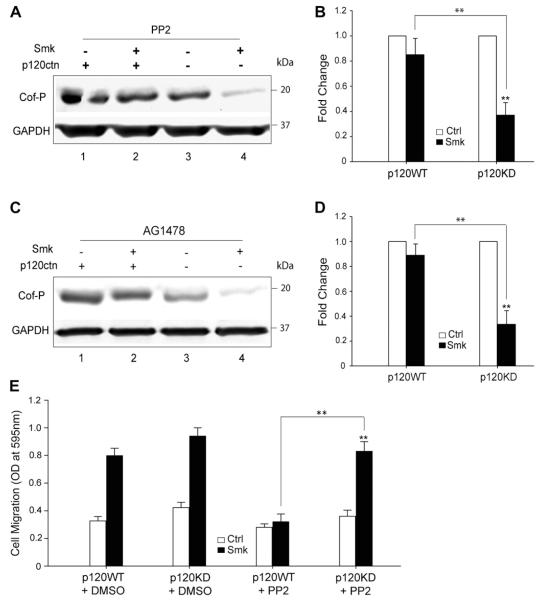
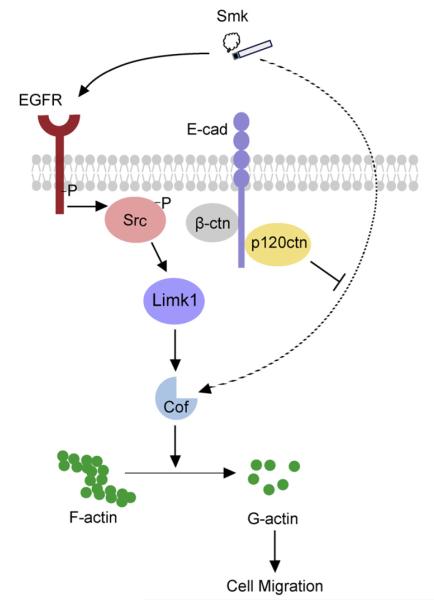
Cigarette smoking has been linked to almost all major types of cancer. Emerging evidence suggests that smoking initiates transformed cell growth and migration by disrupting cell-cell interactions in the polarized mucosal epithelium. Together with other adherens junction proteins, p120-catenin (p120ctn) maintains cell-cell adhesion through its direct interaction with E-cadherin (E-cad). Mislocalization and/or loss of p120ctn have been reported in all lung cancer subtypes and are related to poor prognosis. Here, we showed that p120ctn modulates smoke-induced cell migration via the EGFR/Src-P pathway. Chemical blockade of EGFR/Src signaling inhibited smoke-induced activation of cofilin (an actin severing protein) and promoted cell migration in the presence of p120ctn but had little effect on blocking migration in the absence of p120ctn. These data suggested that smoke-induced cell migration was mediated via an EGFR/Src-dependent signaling pathway in cells that expressed p120ctn, but upon loss of p120ctn, migration continued to occur via an alternative, EGFR/Src-independent pathway. Thus, gradual loss of membrane p120ctn with lung cancer progression may contribute to reduced effectiveness of conventional chemotherapies, such as those directed against EGFR.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures

Similar articles
-
Rac1 and Cdc42 differentially modulate cigarette smoke-induced airway cell migration through p120-catenin-dependent and -independent pathways.Am J Pathol. 2013 Jun;182(6):1986-95. doi: 10.1016/j.ajpath.2013.02.008. Epub 2013 Apr 2. Am J Pathol. 2013. PMID: 23562274 Free PMC article.
-
Pivotal role of MUC1 glycosylation by cigarette smoke in modulating disruption of airway adherens junctions in vitro.J Pathol. 2014 Sep;234(1):60-73. doi: 10.1002/path.4375. Epub 2014 Jul 9. J Pathol. 2014. PMID: 24838315 Free PMC article.
-
Cigarette smoke disrupts the integrity of airway adherens junctions through the aberrant interaction of p120-catenin with the cytoplasmic tail of MUC1.J Pathol. 2013 Jan;229(1):74-86. doi: 10.1002/path.4070. Epub 2012 Sep 28. J Pathol. 2013. PMID: 22833523 Free PMC article.
-
Functions of p120ctn isoforms in cell-cell adhesion and intracellular signaling.Front Biosci (Landmark Ed). 2012 Jan 1;17(5):1669-94. doi: 10.2741/4012. Front Biosci (Landmark Ed). 2012. PMID: 22201829 Review.
-
Functions of p120ctn in development and disease.Front Biosci (Landmark Ed). 2012 Jan 1;17(2):760-83. doi: 10.2741/3956. Front Biosci (Landmark Ed). 2012. PMID: 22201773 Review.
Cited by
-
Jinfu'an Decoction Inhibits Invasion and Metastasis in Human Lung Cancer Cells (H1650) via p120ctn-Mediated Induction and Kaiso.Med Sci Monit. 2018 May 8;24:2878-2886. doi: 10.12659/MSM.909748. Med Sci Monit. 2018. PMID: 29735970 Free PMC article.
-
Rac1 and Cdc42 differentially modulate cigarette smoke-induced airway cell migration through p120-catenin-dependent and -independent pathways.Am J Pathol. 2013 Jun;182(6):1986-95. doi: 10.1016/j.ajpath.2013.02.008. Epub 2013 Apr 2. Am J Pathol. 2013. PMID: 23562274 Free PMC article.
-
Pivotal role of MUC1 glycosylation by cigarette smoke in modulating disruption of airway adherens junctions in vitro.J Pathol. 2014 Sep;234(1):60-73. doi: 10.1002/path.4375. Epub 2014 Jul 9. J Pathol. 2014. PMID: 24838315 Free PMC article.
-
p120-catenin down-regulation and epidermal growth factor receptor overexpression results in a transformed epithelium that mimics esophageal squamous cell carcinoma.Am J Pathol. 2015 Jan;185(1):240-51. doi: 10.1016/j.ajpath.2014.09.008. Am J Pathol. 2015. PMID: 25529795 Free PMC article.
-
Cigarette smoke extract-induced p120-mediated NF-κB activation in human epithelial cells is dependent on the RhoA/ROCK pathway.Sci Rep. 2016 Sep 2;6:23131. doi: 10.1038/srep23131. Sci Rep. 2016. PMID: 27586697 Free PMC article.
References
-
- Shafey O EM, Ross H, Mackay J. The Tobacco Atlas. American Cancer Society; Atlanta (GA): 2009.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
