

Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases
- PMID: 22120714
- PMCID: PMC3378484
- DOI: 10.1038/onc.2011.532
Regulation of lipid binding underlies the activation mechanism of class IA PI3-kinases
Abstract
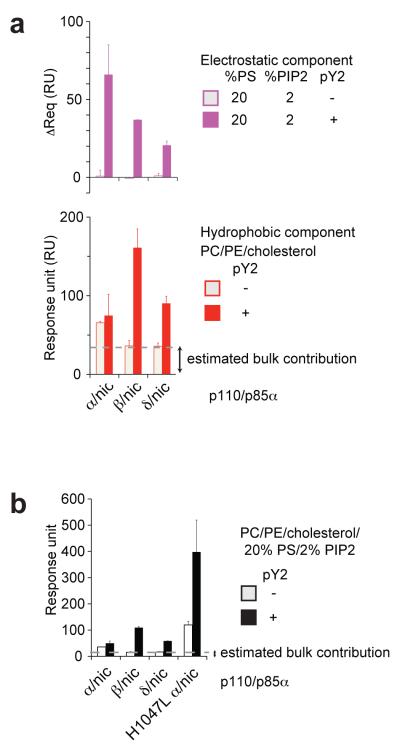
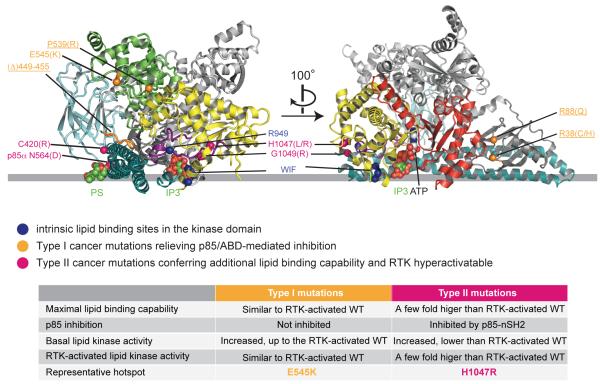
Somatic missense mutations in PIK3CA, which encodes the p110α catalytic subunit of phosphoinositide 3-kinases, occur frequently in human cancers. Activating mutations spread across multiple domains, some of which are located at inhibitory contact sites formed with the regulatory subunit p85α. PIK3R1, which encodes p85α, also has activating somatic mutations. We find a strong correlation between lipid kinase and lipid-binding activities for both wild-type (WT) and a representative set of oncogenic mutant complexes of p110α/p85α. Lipid binding involves both electrostatic and hydrophobic interactions. Activation caused by a phosphorylated receptor tyrosine kinase (RTK) peptide binding to the p85α N-terminal SH2 domain (nSH2) induces lipid binding. This depends on the polybasic activation loop as well as a conserved hydrophobic motif in the C-terminal region of the kinase domain. The hotspot E545K mutant largely mimics the activated WT p110α. It shows the highest basal activity and lipid binding, and is not significantly activated by an RTK phosphopeptide. Both the hotspot H1047R mutant and rare mutations (C420R, M1043I, H1047L, G1049R and p85α-N564D) also show increased basal kinase activities and lipid binding. However, their activities are further enhanced by an RTK phosphopeptide to levels markedly exceeding that of activated WT p110α. Phosphopeptide binding to p110β/p85α and p110δ/p85α complexes also induces their lipid binding. We present a crystal structure of WT p110α complexed with the p85α inter-SH2 domain and the inhibitor PIK-108. Additional to the ATP-binding pocket, an unexpected, second PIK-108 binding site is observed in the kinase C-lobe. We show a global conformational change in p110α consistent with allosteric regulation of the kinase domain by nSH2. These findings broaden our understanding of the differential biological outputs exhibited by distinct types of mutations regarding growth factor dependence, and suggest a two-tier classification scheme relating p110α and p85α mutations with signalling potential.
Figures
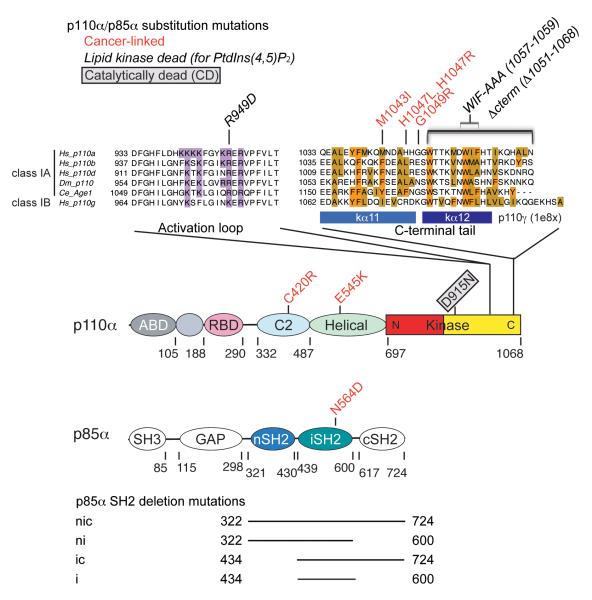
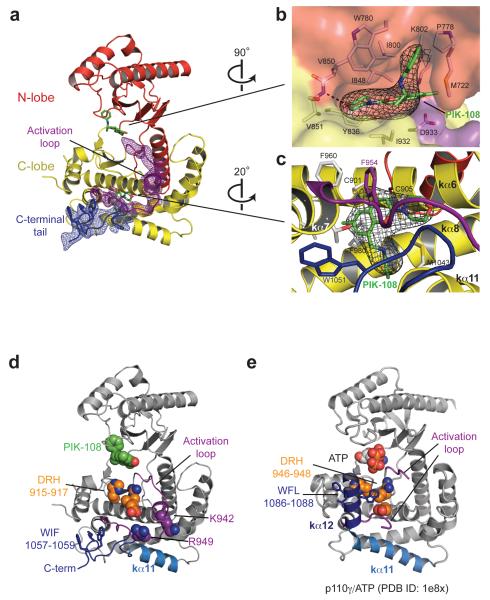
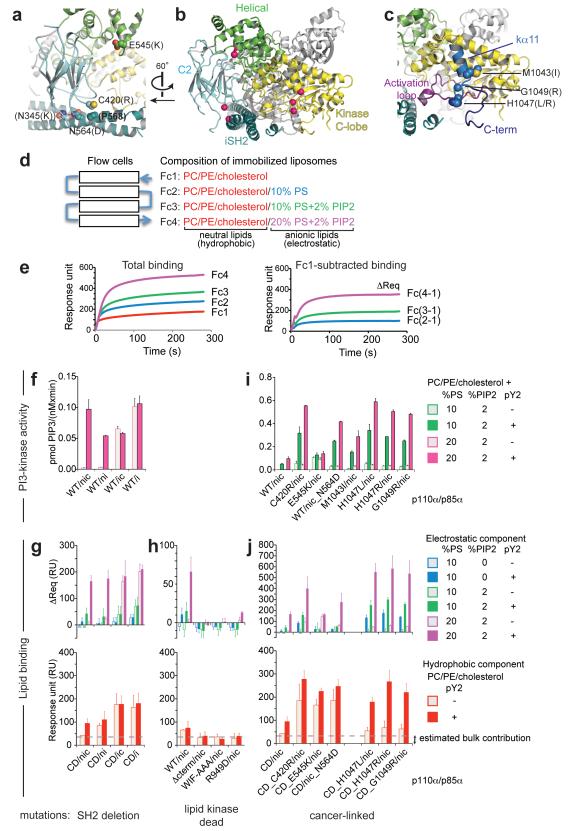
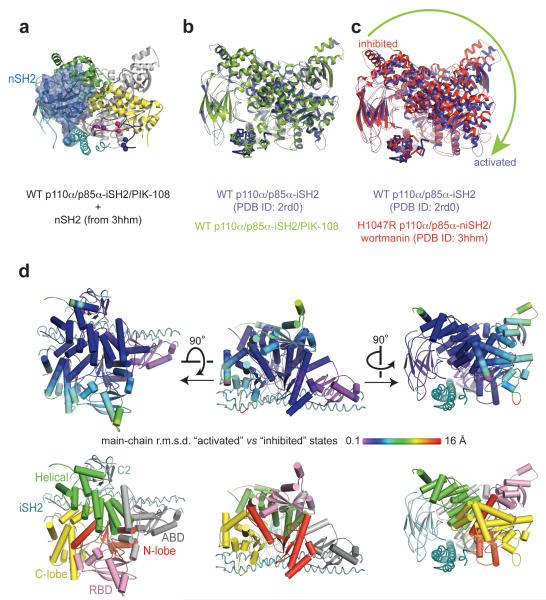
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
