

A meckelin-filamin A interaction mediates ciliogenesis
- PMID: 22121117
- PMCID: PMC3284117
- DOI: 10.1093/hmg/ddr557
A meckelin-filamin A interaction mediates ciliogenesis
Abstract
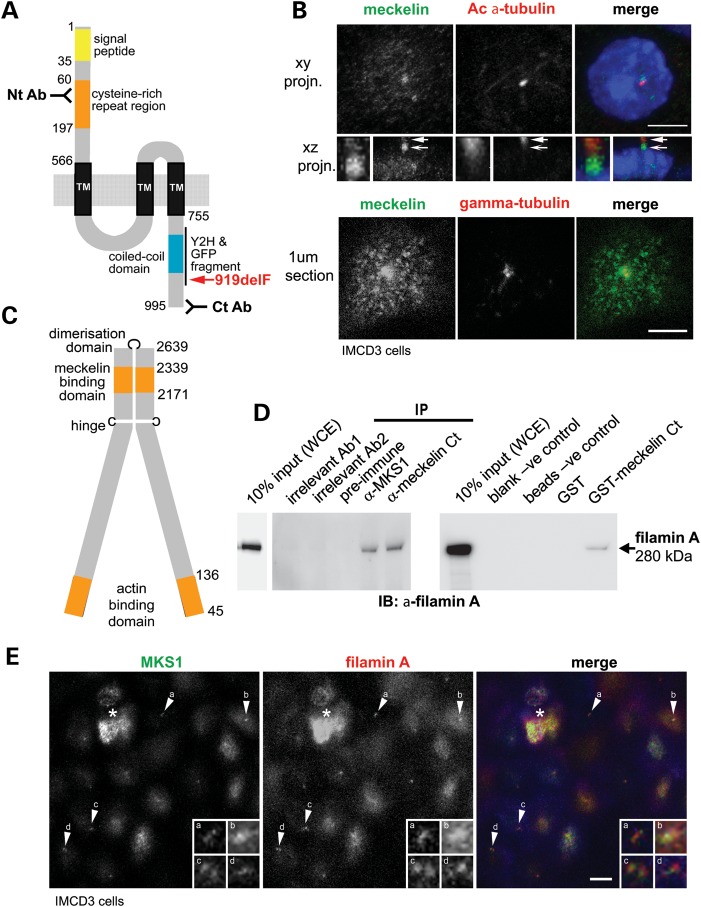
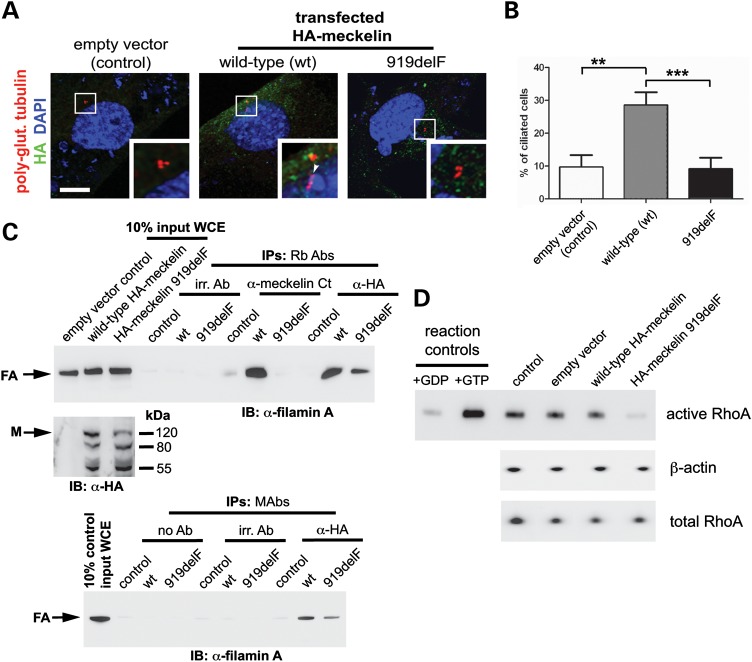
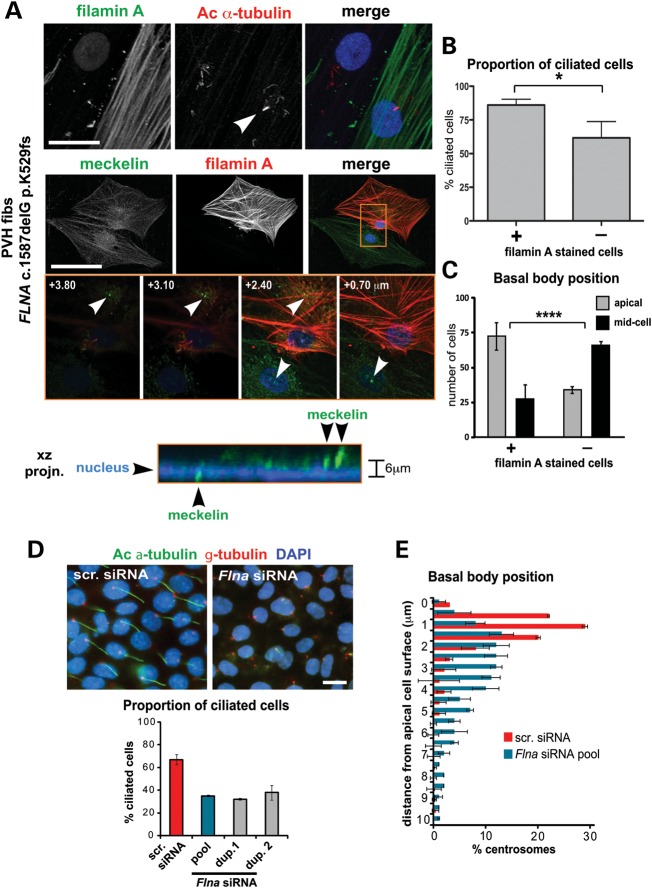
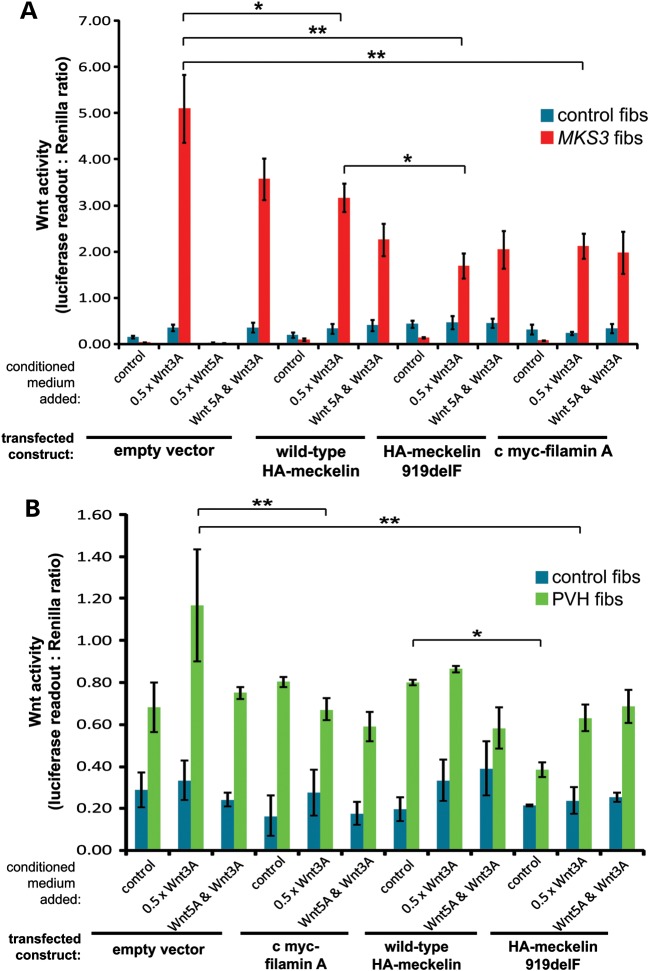
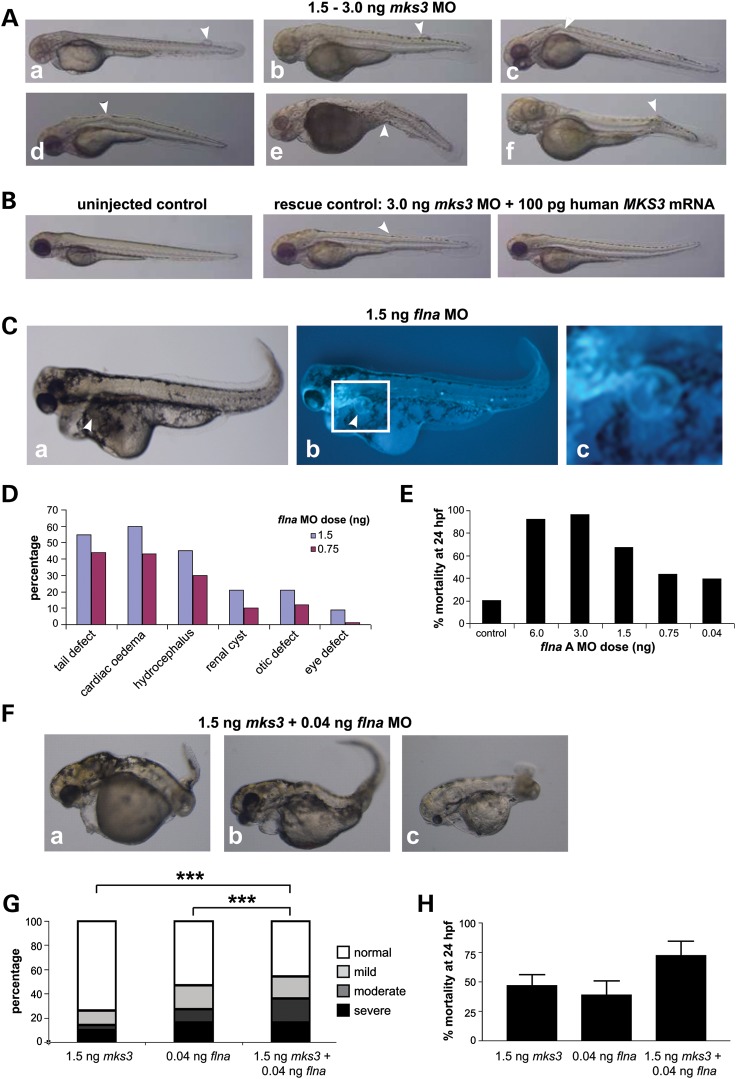
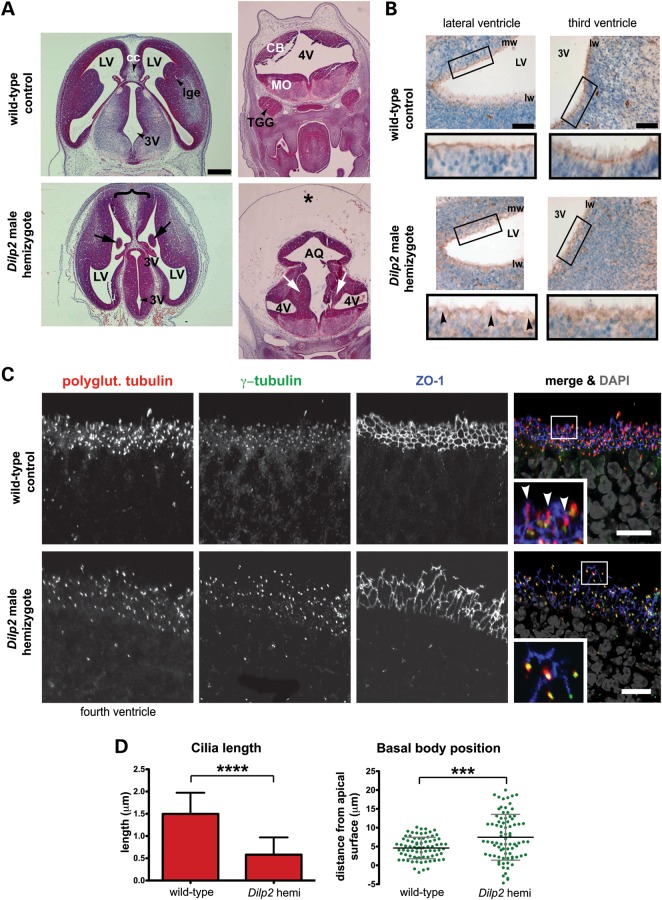
MKS3, encoding the transmembrane receptor meckelin, is mutated in Meckel-Gruber syndrome (MKS), an autosomal-recessive ciliopathy. Meckelin localizes to the primary cilium, basal body and elsewhere within the cell. Here, we found that the cytoplasmic domain of meckelin directly interacts with the actin-binding protein filamin A, potentially at the apical cell surface associated with the basal body. Mutations in FLNA, the gene for filamin A, cause periventricular heterotopias. We identified a single consanguineous patient with an MKS-like ciliopathy that presented with both MKS and cerebellar heterotopia, caused by an unusual in-frame deletion mutation in the meckelin C-terminus at the region of interaction with filamin A. We modelled this mutation and found it to abrogate the meckelin-filamin A interaction. Furthermore, we found that loss of filamin A by siRNA knockdown, in patient cells, and in tissues from Flna(Dilp2) null mouse embryos results in cellular phenotypes identical to those caused by meckelin loss, namely basal body positioning and ciliogenesis defects. In addition, morpholino knockdown of flna in zebrafish embryos significantly increases the frequency of dysmorphology and severity of ciliopathy developmental defects caused by mks3 knockdown. Our results suggest that meckelin forms a functional complex with filamin A that is disrupted in MKS and causes defects in neuronal migration and Wnt signalling. Furthermore, filamin A has a crucial role in the normal processes of ciliogenesis and basal body positioning. Concurrent with these processes, the meckelin-filamin A signalling axis may be a key regulator in maintaining correct, normal levels of Wnt signalling.
Figures
References
-
- Baala L., Audollent S., Martinovic J., Ozilou C., Babron M.C., Sivanandamoorthy S., Saunier S., Salomon R., Gonzales M., Rattenberry E., et al. Pleiotropic effects of CEP290 (NPHP6) mutations extend to Meckel syndrome. Am. J. Hum. Genet. 2007;81:170–179. doi:, doi:10.1086/519494. - DOI - PMC - PubMed
-
- Kyttala M., Tallila J., Salonen R., Kopra O., Kohlschmidt N., Paavola-Sakki P., Peltonen L., Kestila M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat. Genet. 2006;38:155–157. doi:10.1016/S0022-0981(03)00271-5, doi:10.1038/ng1714. - DOI - DOI - PubMed
-
- Smith U.M., Consugar M., Tee L.J., McKee B.M., Maina E.N., Whelan S., Morgan N.V., Goranson E., Gissen P., Lilliquist S., et al. The transmembrane protein meckelin (MKS3) is mutated in Meckel–Gruber syndrome and the wpk rat. Nat. Genet. 2006;38:191–196. doi:10.1080/00222939000770621, doi:10.1038/ng1713. - DOI - DOI - PubMed
-
- Valente E.M., Logan C.V., Mougou-Zerelli S., Lee J.H., Silhavy J.L., Brancati F., Iannicelli M., Travaglini L., Romani S., Illi B., et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet. 2010;42:619–625. doi:, doi:10.1038/ng.594. - DOI - PMC - PubMed
-
- Roume J., Genin E., Cormier-Daire V., Ma H.W., Mehaye B., Attie T., Razavi-Encha F., Fallet-Bianco C., Buenerd A., Clerget-Darpoux F., et al. A gene for Meckel syndrome maps to chromosome 11q13. Am. J. Hum. Genet. 1998;63:1095–1101. doi:10.1086/302062. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
