

Cell death and infection: a double-edged sword for host and pathogen survival
- PMID: 22123830
- PMCID: PMC3241725
- DOI: 10.1083/jcb.201108081
Cell death and infection: a double-edged sword for host and pathogen survival
Abstract
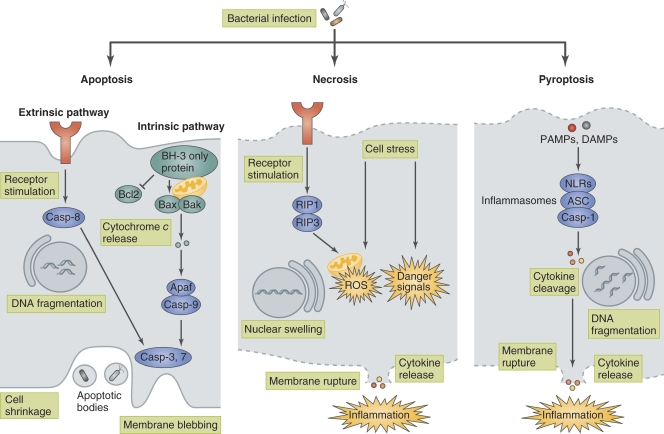
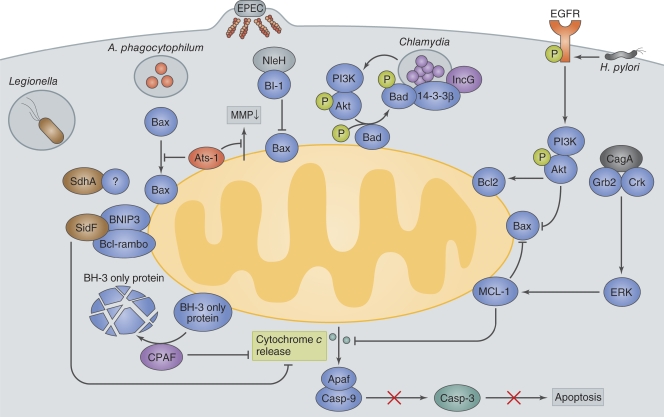
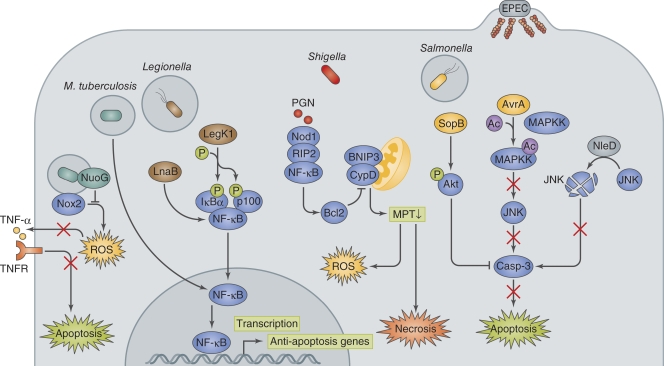
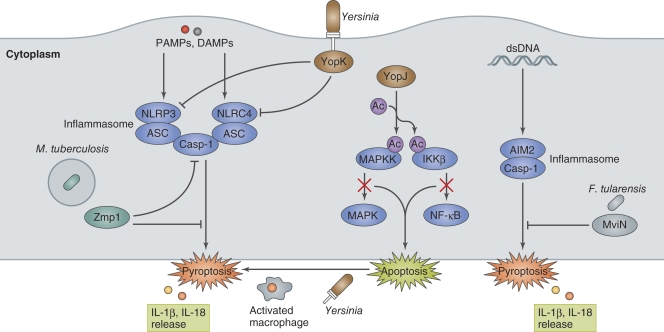
Host cell death is an intrinsic immune defense mechanism in response to microbial infection. However, bacterial pathogens use many strategies to manipulate the host cell death and survival pathways to enhance their replication and survival. This manipulation is quite intricate, with pathogens often suppressing cell death to allow replication and then promoting it for dissemination. Frequently, these effects are exerted through modulation of the mitochondrial pro-death, NF-κB-dependent pro-survival, and inflammasome-dependent host cell death pathways during infection. Understanding the molecular details by which bacterial pathogens manipulate cell death pathways will provide insight into new therapeutic approaches to control infection.
Figures
References
-
- Amer A., Franchi L., Kanneganti T.D., Body-Malapel M., Ozören N., Brady G., Meshinchi S., Jagirdar R., Gewirtz A., Akira S., Núñez G. 2006. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J. Biol. Chem. 281:35217–35223 10.1074/jbc.M604933200 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
