

Haplotype variation and genotype imputation in African populations
- PMID: 22125220
- PMCID: PMC3568705
- DOI: 10.1002/gepi.20626
Haplotype variation and genotype imputation in African populations
Abstract
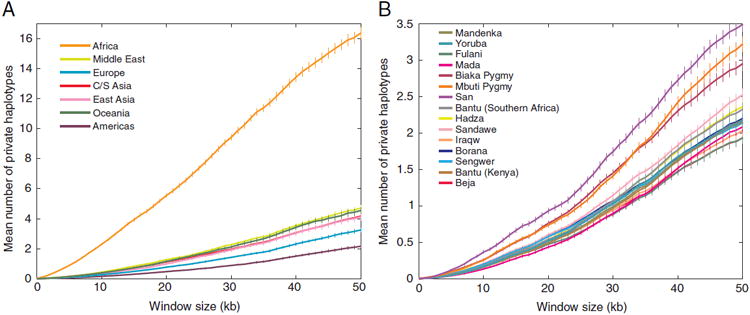
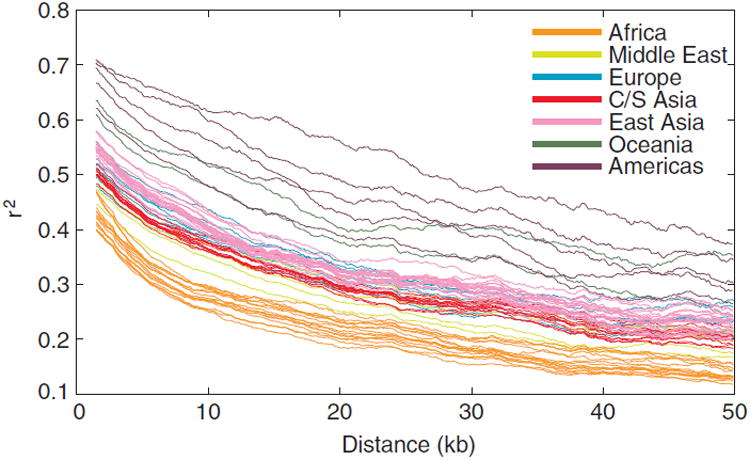
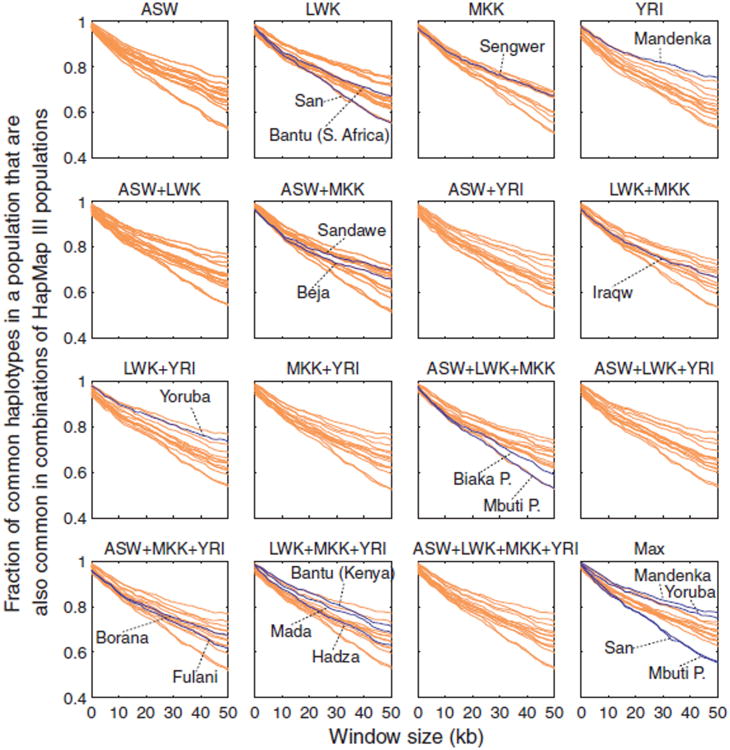
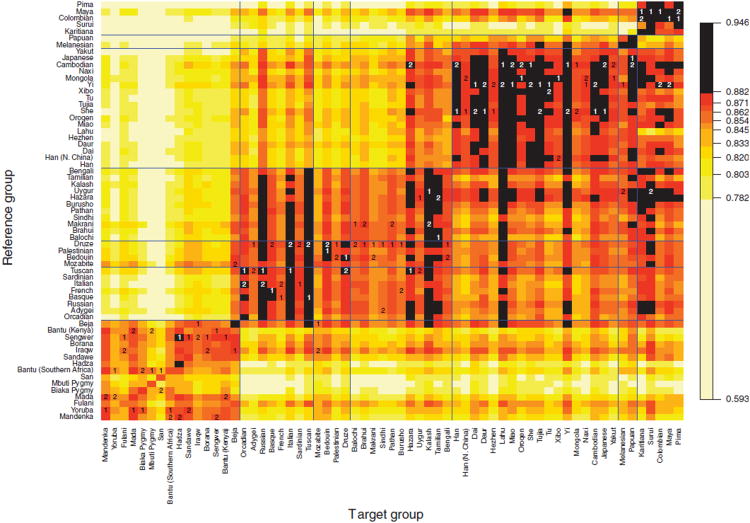
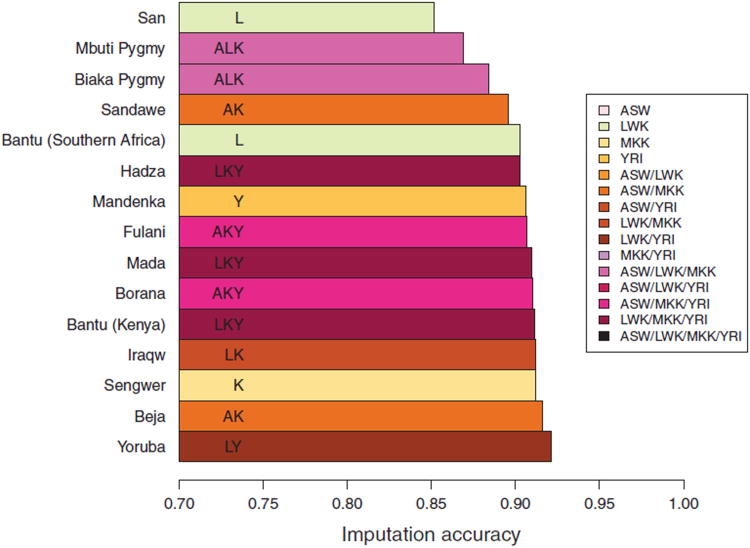
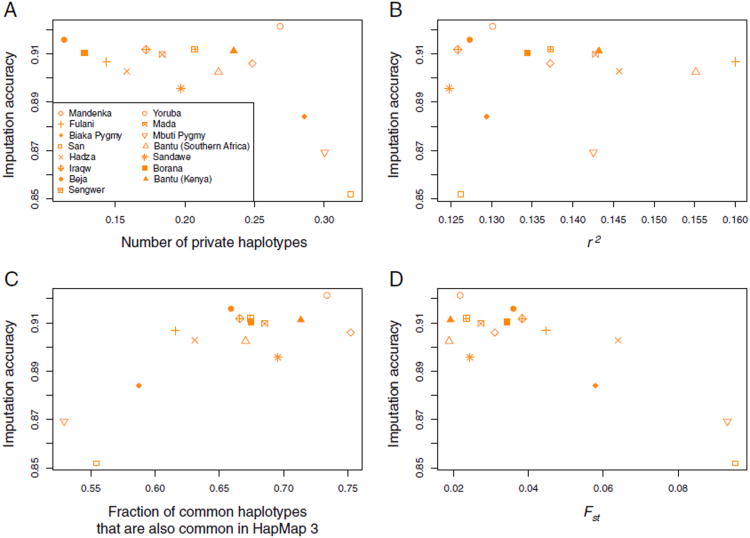
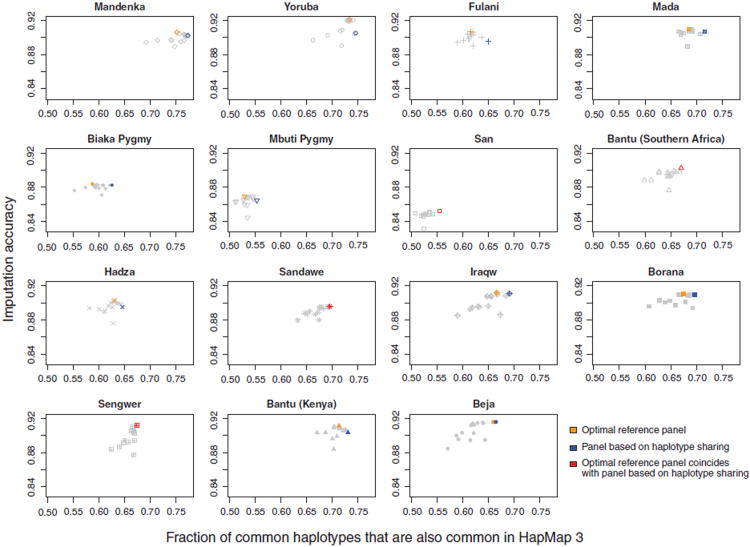
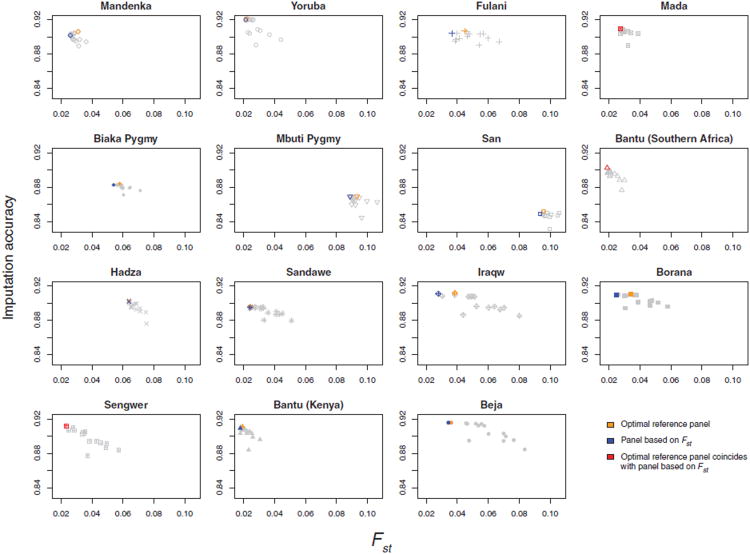
Sub-Saharan Africa has been identified as the part of the world with the greatest human genetic diversity. This high level of diversity causes difficulties for genome-wide association (GWA) studies in African populations-for example, by reducing the accuracy of genotype imputation in African populations compared to non-African populations. Here, we investigate haplotype variation and imputation in Africa, using 253 unrelated individuals from 15 Sub-Saharan African populations. We identify the populations that provide the greatest potential for serving as reference panels for imputing genotypes in the remaining groups. Considering reference panels comprising samples of recent African descent in Phase 3 of the HapMap Project, we identify mixtures of reference groups that produce the maximal imputation accuracy in each of the sampled populations. We find that optimal HapMap mixtures and maximal imputation accuracies identified in detailed tests of imputation procedures can instead be predicted by using simple summary statistics that measure relationships between the pattern of genetic variation in a target population and the patterns in potential reference panels. Our results provide an empirical basis for facilitating the selection of reference panels in GWA studies of diverse human populations, especially those of African ancestry.
© 2011 Wiley Periodicals, Inc.
Figures
References
-
- Bowcock AM, Ruiz-Linares A, Tomfohrde J, Minch E, Kidd JR, Cavalli-Sforza LL. High resolution of human evolutionary trees with polymorphic microsatellites. Nature. 1994;368:455–457. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
