

Pitfalls of merging GWAS data: lessons learned in the eMERGE network and quality control procedures to maintain high data quality
- PMID: 22125226
- PMCID: PMC3592376
- DOI: 10.1002/gepi.20639
Pitfalls of merging GWAS data: lessons learned in the eMERGE network and quality control procedures to maintain high data quality
Abstract
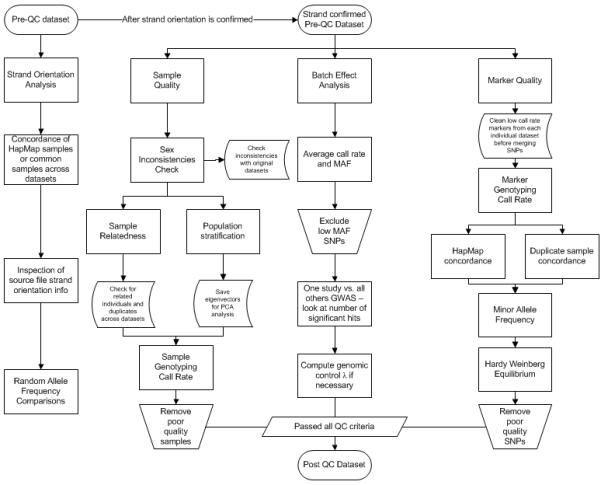
Genome-wide association studies (GWAS) are a useful approach in the study of the genetic components of complex phenotypes. Aside from large cohorts, GWAS have generally been limited to the study of one or a few diseases or traits. The emergence of biobanks linked to electronic medical records (EMRs) allows the efficient reuse of genetic data to yield meaningful genotype-phenotype associations for multiple phenotypes or traits. Phase I of the electronic MEdical Records and GEnomics (eMERGE-I) Network is a National Human Genome Research Institute-supported consortium composed of five sites to perform various genetic association studies using DNA repositories and EMR systems. Each eMERGE site has developed EMR-based algorithms to comprise a core set of 14 phenotypes for extraction of study samples from each site's DNA repository. Each eMERGE site selected samples for a specific phenotype, and these samples were genotyped at either the Broad Institute or at the Center for Inherited Disease Research using the Illumina Infinium BeadChip technology. In all, approximately 17,000 samples from across the five sites were genotyped. A unified quality control (QC) pipeline was developed by the eMERGE Genomics Working Group and used to ensure thorough cleaning of the data. This process includes examination of sample and marker quality and various batch effects. Upon completion of the genotyping and QC analyses for each site's primary study, eMERGE Coordinating Center merged the datasets from all five sites. This larger merged dataset reentered the established eMERGE QC pipeline. Based on lessons learned during the process, additional analyses and QC checkpoints were added to the pipeline to ensure proper merging. Here, we explore the challenges associated with combining datasets from different genotyping centers and describe the expansion to eMERGE QC pipeline for merged datasets. These additional steps will be useful as the eMERGE project expands to include additional sites in eMERGE-II, and also serve as a starting point for investigators merging multiple genotype datasets accessible through the National Center for Biotechnology Information in the database of Genotypes and Phenotypes. Our experience demonstrates that merging multiple datasets after additional QC can be an efficient use of genotype data despite new challenges that appear in the process.
© 2011 Wiley Periodicals, Inc.
Figures
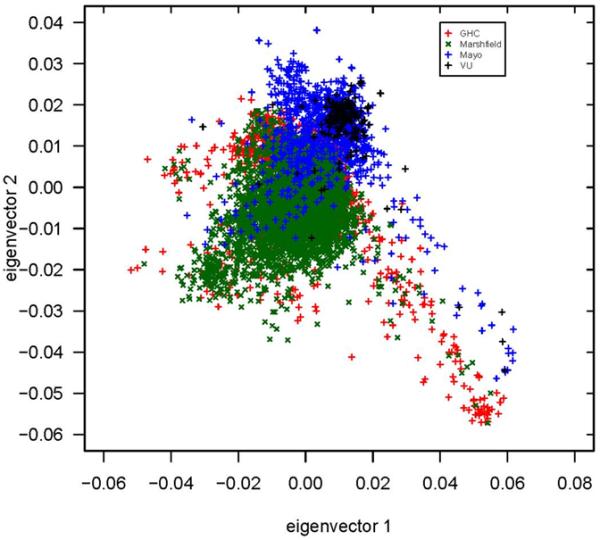
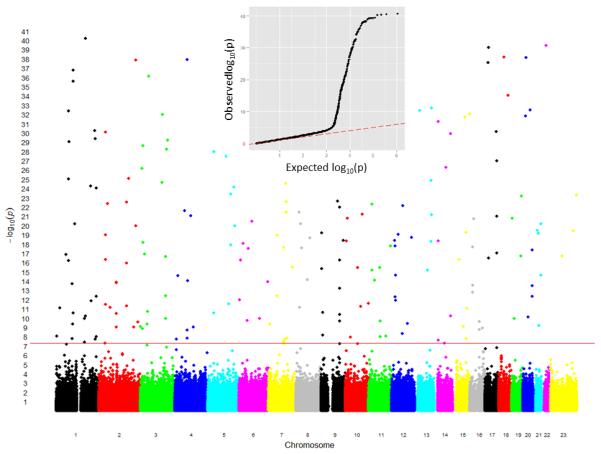
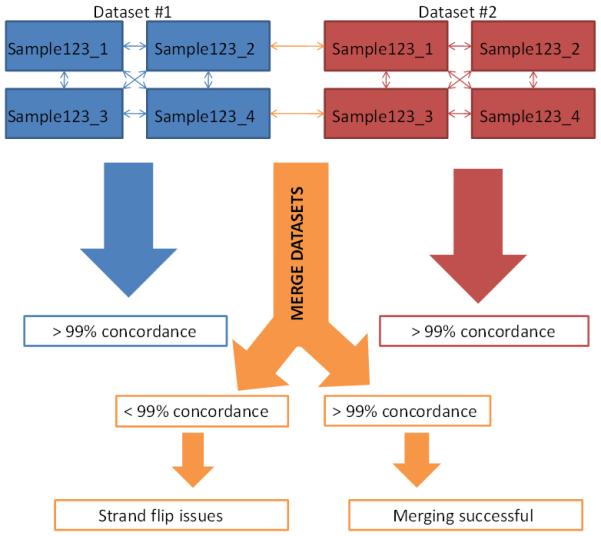
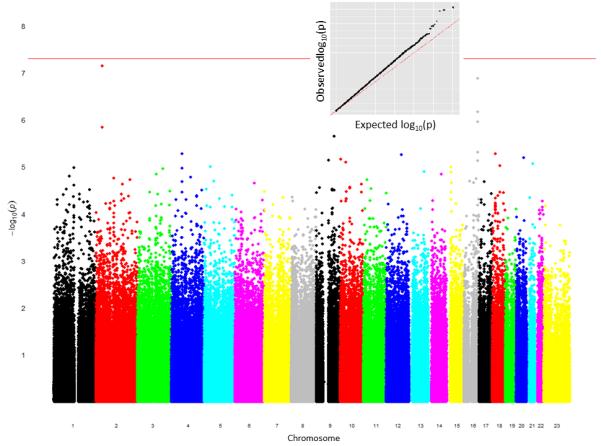
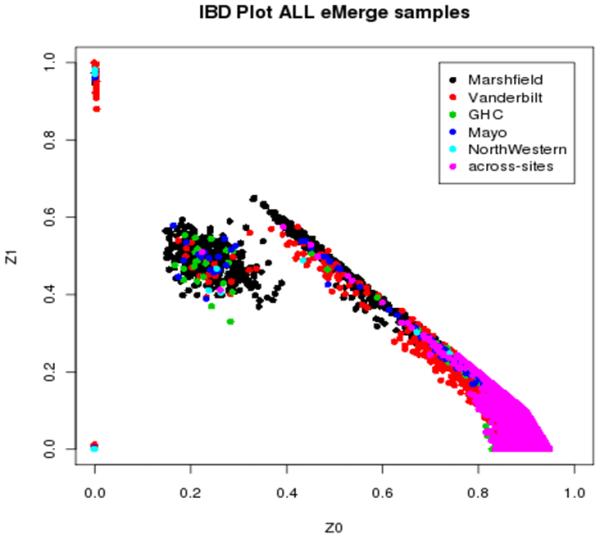
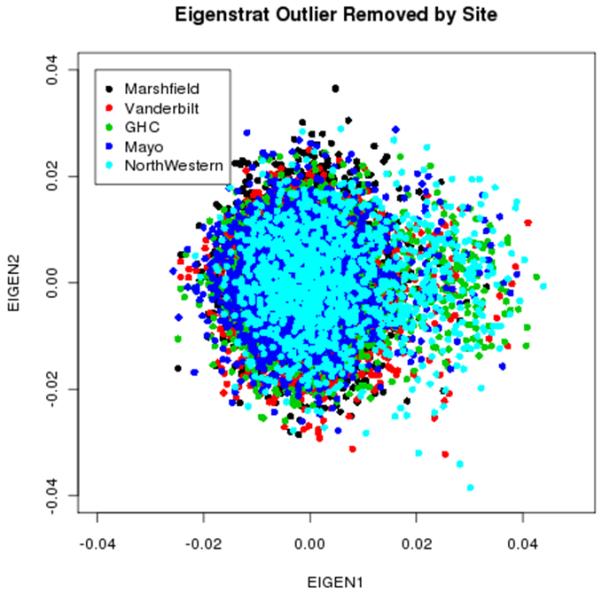
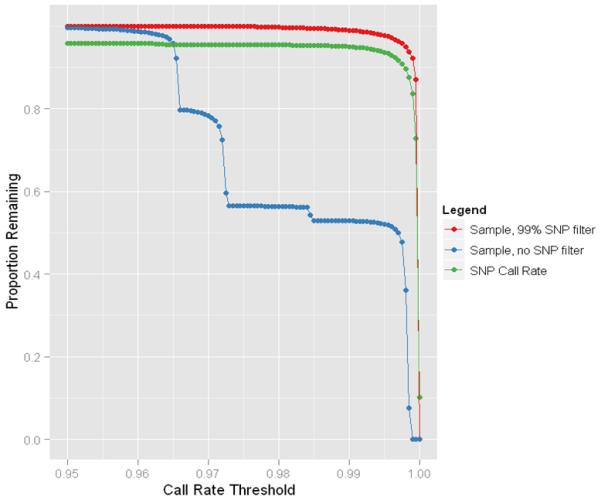
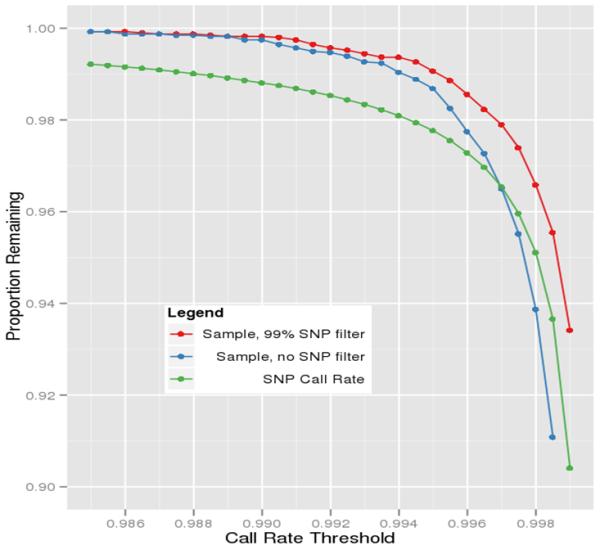
References
-
- Manolio TA. Genomewide association studies and assessment of the risk of disease. N. Engl. J. Med. 2010;363:166–176. - PubMed
-
- Pritchard JK, Cox NJ. The allelic architecture of human disease genes: common disease-common variant…or not? Hum. Mol. Genet. 2002;11:2417–2423. - PubMed
-
- Reich DE, Lander ES. On the allelic spectrum of human disease. Trends Genet. 2001;17:502–510. - PubMed
Publication types
MeSH terms
Grants and funding
- U01HG004438/HG/NHGRI NIH HHS/United States
- T32 GM080178/GM/NIGMS NIH HHS/United States
- U01HG004610/HG/NHGRI NIH HHS/United States
- U01 HG004603/HG/NHGRI NIH HHS/United States
- U01HG04603/HG/NHGRI NIH HHS/United States
- U01HG004609/HG/NHGRI NIH HHS/United States
- U01 HG004609/HG/NHGRI NIH HHS/United States
- U01 HG004599/HG/NHGRI NIH HHS/United States
- U01HG004608/HG/NHGRI NIH HHS/United States
- U01 HG004608/HG/NHGRI NIH HHS/United States
- U01 HG006375/HG/NHGRI NIH HHS/United States
- U01 HG004438/HG/NHGRI NIH HHS/United States
- U01HG04599/HG/NHGRI NIH HHS/United States
- ImNIH/Intramural NIH HHS/United States
- R01 LM010040/LM/NLM NIH HHS/United States
- U01 HG004610/HG/NHGRI NIH HHS/United States
- R01LM010040/LM/NLM NIH HHS/United States
LinkOut - more resources
Full Text Sources
