

Heritable epigenetic variation among maize inbreds
- PMID: 22125494
- PMCID: PMC3219600
- DOI: 10.1371/journal.pgen.1002372
Heritable epigenetic variation among maize inbreds
Abstract
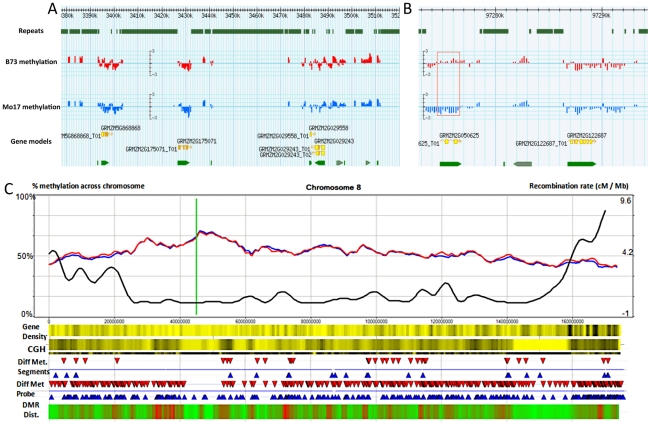
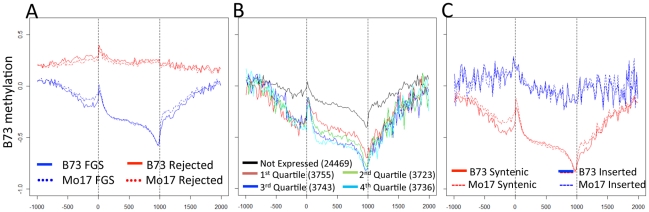
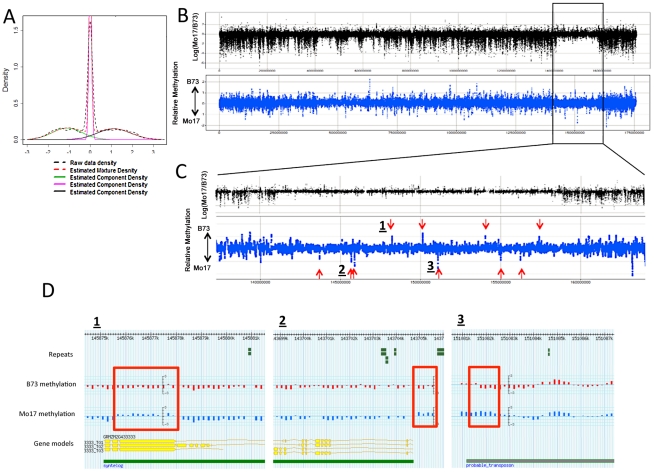
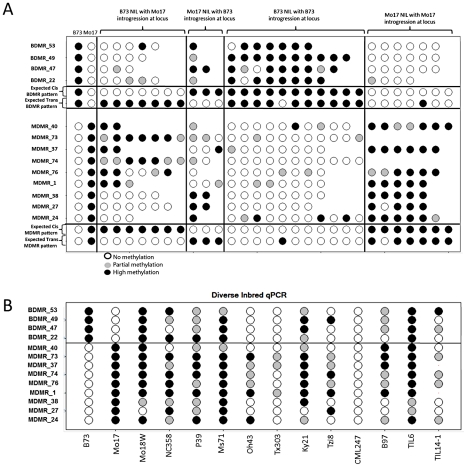
Epigenetic variation describes heritable differences that are not attributable to changes in DNA sequence. There is the potential for pure epigenetic variation that occurs in the absence of any genetic change or for more complex situations that involve both genetic and epigenetic differences. Methylation of cytosine residues provides one mechanism for the inheritance of epigenetic information. A genome-wide profiling of DNA methylation in two different genotypes of Zea mays (ssp. mays), an organism with a complex genome of interspersed genes and repetitive elements, allowed the identification and characterization of examples of natural epigenetic variation. The distribution of DNA methylation was profiled using immunoprecipitation of methylated DNA followed by hybridization to a high-density tiling microarray. The comparison of the DNA methylation levels in the two genotypes, B73 and Mo17, allowed for the identification of approximately 700 differentially methylated regions (DMRs). Several of these DMRs occur in genomic regions that are apparently identical by descent in B73 and Mo17 suggesting that they may be examples of pure epigenetic variation. The methylation levels of the DMRs were further studied in a panel of near-isogenic lines to evaluate the stable inheritance of the methylation levels and to assess the contribution of cis- and trans- acting information to natural epigenetic variation. The majority of DMRs that occur in genomic regions without genetic variation are controlled by cis-acting differences and exhibit relatively stable inheritance. This study provides evidence for naturally occurring epigenetic variation in maize, including examples of pure epigenetic variation that is not conditioned by genetic differences. The epigenetic differences are variable within maize populations and exhibit relatively stable trans-generational inheritance. The detected examples of epigenetic variation, including some without tightly linked genetic variation, may contribute to complex trait variation.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Bird A. Perceptions of epigenetics. Nature. 2007;447(7143):396–398. - PubMed
-
- Chan SW, Henderson IR, Jacobsen SE. Gardening the genome: DNA methylation in arabidopsis thaliana. Nat Rev Genet. 2005;6:351–360. - PubMed
-
- Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007;8:272–285. 10.1038/nrg2072. - PubMed
-
- Rasmusson DC, Phillips RL. Plant breeding progress and genetic diversity from de novo variation and elevated epistasis. Crop Sci. 1997;37:303–310.
-
- Richards EJ. Inherited epigenetic variation–revisiting soft inheritance. Nat Rev Genet. 2006;7:395–401. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
