

In Vivo Modulation of Dendritic Cells by Engineered Materials: Towards New Cancer Vaccines
- PMID: 22125572
- PMCID: PMC3224090
- DOI: 10.1016/j.nantod.2011.08.005
In Vivo Modulation of Dendritic Cells by Engineered Materials: Towards New Cancer Vaccines
Abstract
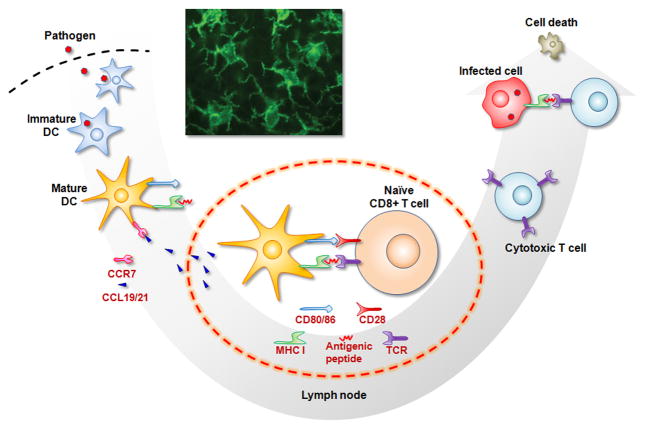
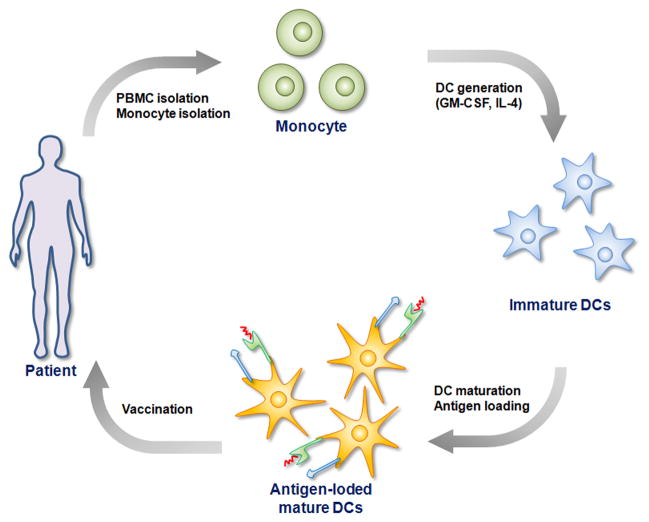
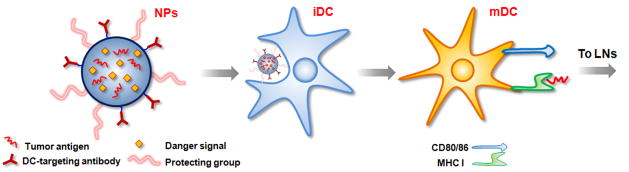
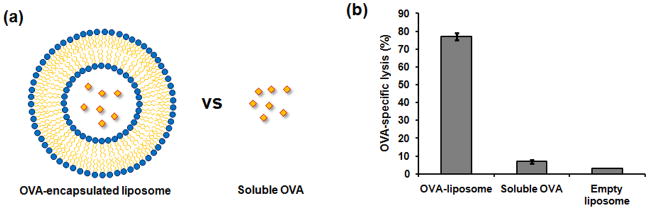
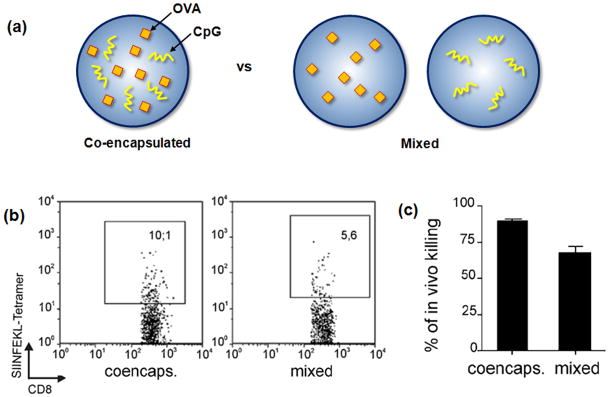
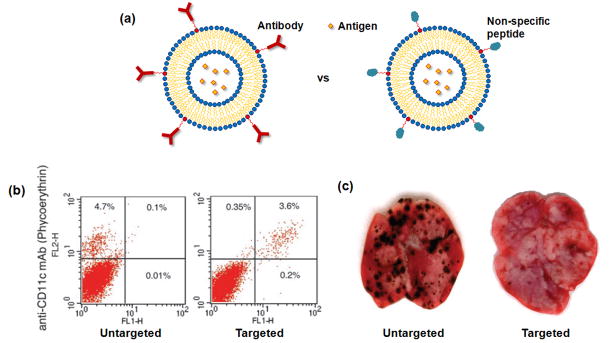
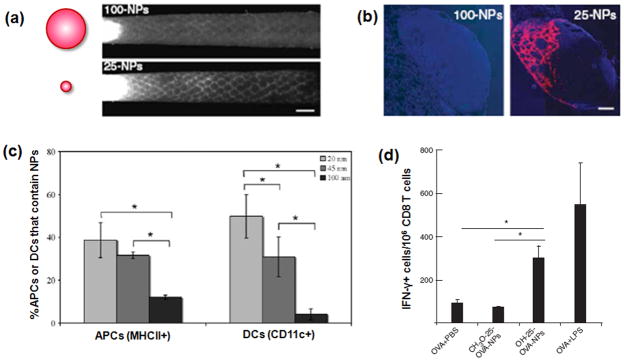
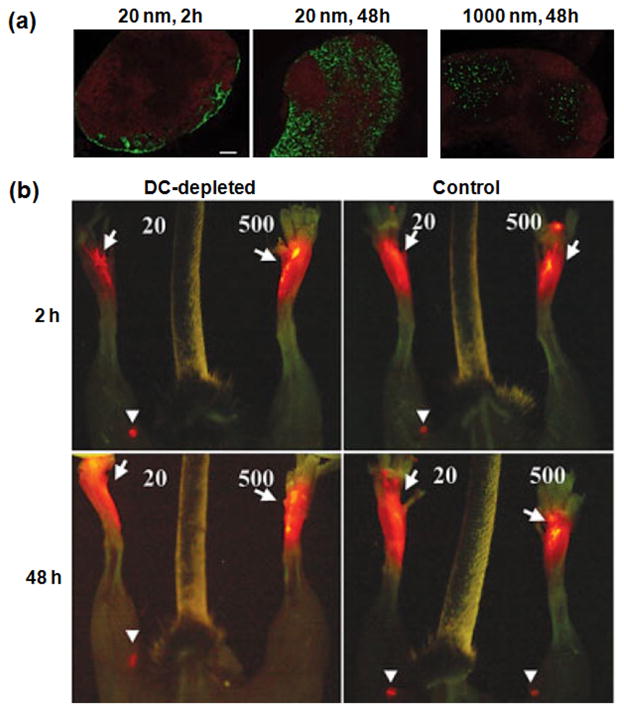
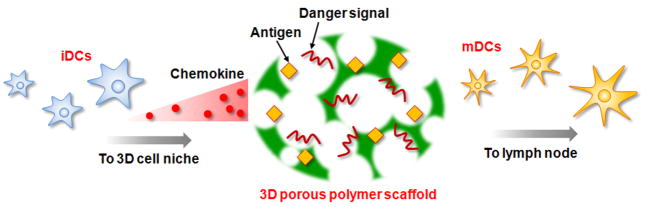
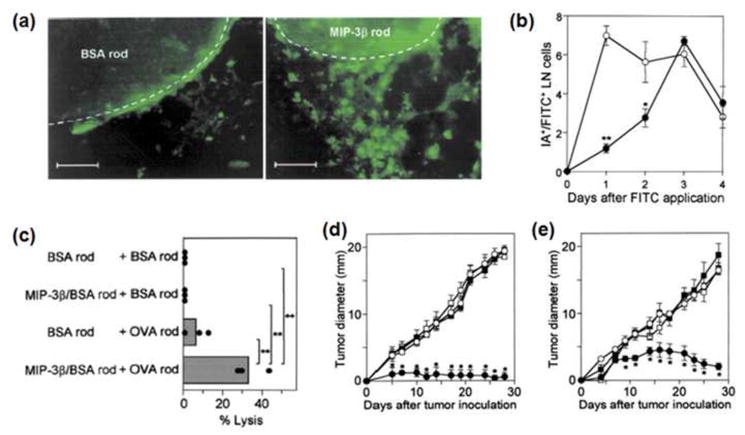
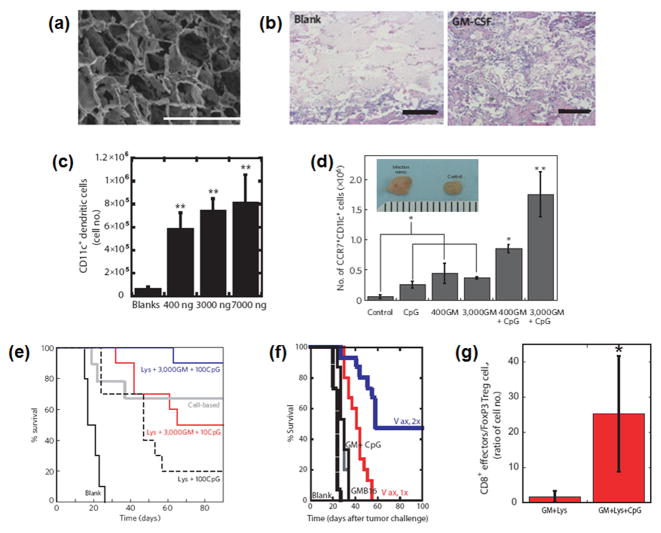
Therapeutic cancer vaccines are emerging as novel and potent approaches to treat cancer. These vaccines enhance the body's immune response to cancerous cells, and dendritic cells (DCs), an initiator of adaptive immunity, are a key cell type targeted by these strategies. Current DC-based cancer vaccines are based on ex vivo manipulation of the cells following their isolation from the patient, followed by reintroduction to the patient, but this approach has many limitations in practical cancer treatment. However, recent progress in materials science has allowed the design and fabrication of physically and chemically functionalized materials platforms that can specifically target DCs in the body. These materials, through their in vivo modulation of DCs, have tremendous potentials as new cancer therapies. Nanoparticles, which are several orders of magnitude smaller than DCs, can efficiently deliver antigen and danger signals to these cells through passive or active targeting. Three-dimensional biomaterials, with sizes several orders of magnitude larger than DCs, create microenvironments that allow the effective recruitment and programming of these cells, and can be used as local depots of nanoparticles targeting resident DCs. Both material strategies have shown potential in promoting antigen-specific T cell responses of magnitudes relevant to treating cancer.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources