

Transforming growth factor β₁ oppositely regulates the hypertrophic and contractile response to β-adrenergic stimulation in the heart
- PMID: 22125598
- PMCID: PMC3219639
- DOI: 10.1371/journal.pone.0026628
Transforming growth factor β₁ oppositely regulates the hypertrophic and contractile response to β-adrenergic stimulation in the heart
Abstract
Background: Neuroendocrine activation and local mediators such as transforming growth factor-β₁ (TGF-β₁) contribute to the pathobiology of cardiac hypertrophy and failure, but the underlying mechanisms are incompletely understood. We aimed to characterize the functional network involving TGF-β₁, the renin-angiotensin system, and the β-adrenergic system in the heart.
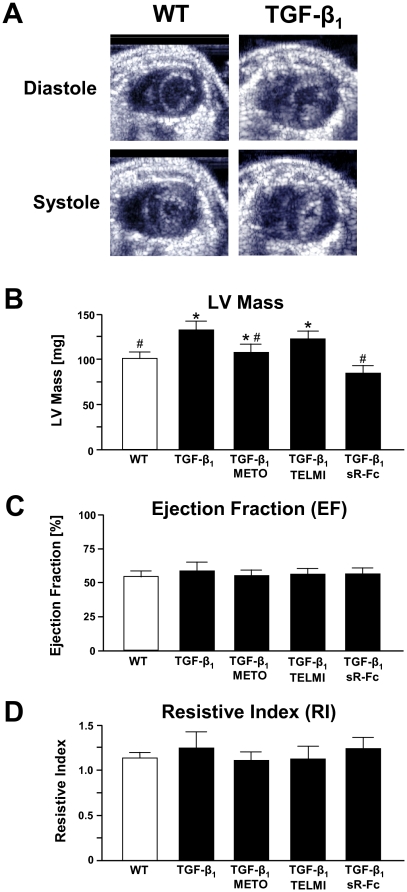
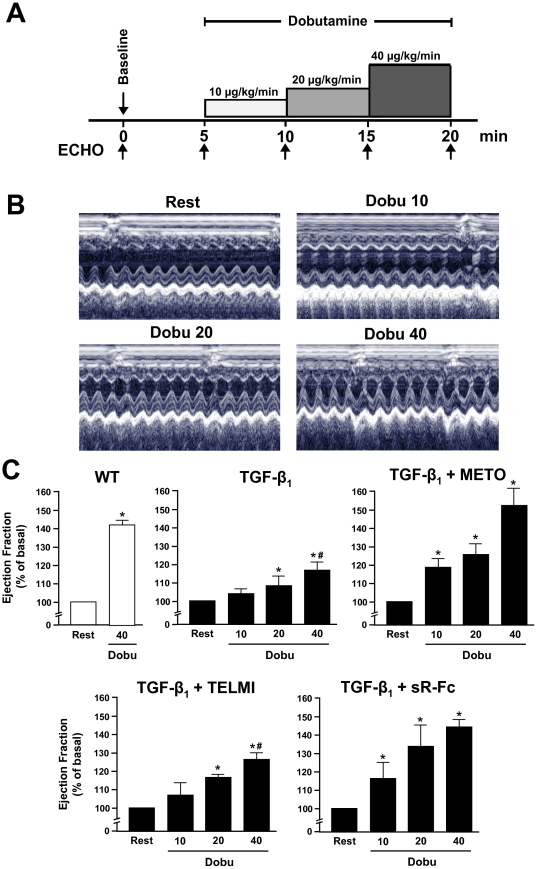
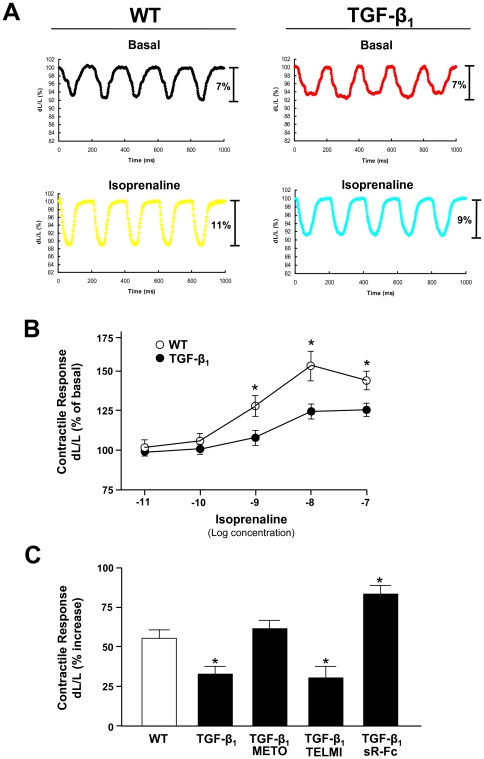
Methods: Transgenic mice overexpressing TGF-β₁ (TGF-β₁-Tg) were treated with a β-blocker, an AT₁-receptor antagonist, or a TGF-β-antagonist (sTGFβR-Fc), were morphologically characterized. Contractile function was assessed by dobutamine stress echocardiography in vivo and isolated myocytes in vitro. Functional alterations were related to regulators of cardiac energy metabolism.
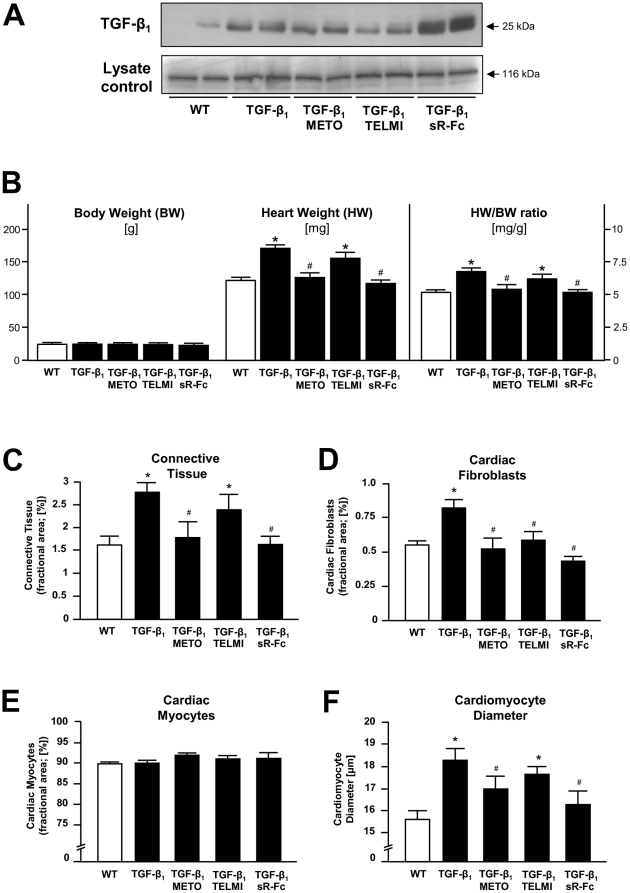
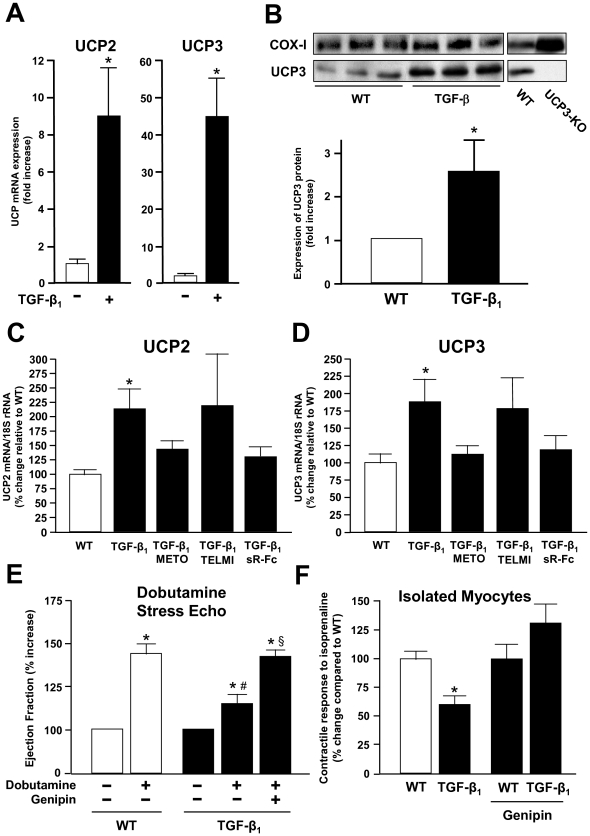
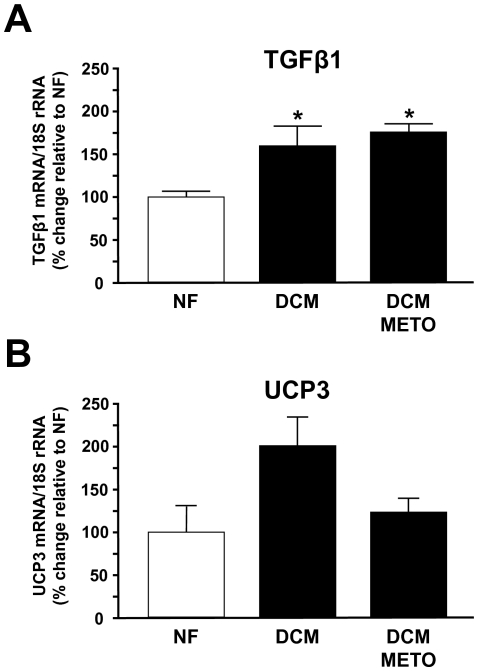
Results: Compared to wild-type controls, TGF-β₁-Tg mice displayed an increased heart-to-body-weight ratio involving both fibrosis and myocyte hypertrophy. TGF-β₁ overexpression increased the hypertrophic responsiveness to β-adrenergic stimulation. In contrast, the inotropic response to β-adrenergic stimulation was diminished in TGF-β₁-Tg mice, albeit unchanged basal contractility. Treatment with sTGF-βR-Fc completely prevented the cardiac phenotype in transgenic mice. Chronic β-blocker treatment also prevented hypertrophy and ANF induction by isoprenaline, and restored the inotropic response to β-adrenergic stimulation without affecting TGF-β₁ levels, whereas AT₁-receptor blockade had no effect. The impaired contractile reserve in TGF-β₁-Tg mice was accompanied by an upregulation of mitochondrial uncoupling proteins (UCPs) which was reversed by β-adrenoceptor blockade. UCP-inhibition restored the contractile response to β-adrenoceptor stimulation in vitro and in vivo. Finally, cardiac TGF-β₁ and UCP expression were elevated in heart failure in humans, and UCP--but not TGF-β₁--was downregulated by β-blocker treatment.
Conclusions: Our data support the concept that TGF-β₁ acts downstream of angiotensin II in cardiomyocytes, and furthermore, highlight the critical role of the β-adrenergic system in TGF-β₁-induced cardiac phenotype. Our data indicate for the first time, that TGF-β₁ directly influences mitochondrial energy metabolism by regulating UCP3 expression. β-blockers may act beneficially by normalizing regulatory mechanisms of cellular hypertrophy and energy metabolism.
Conflict of interest statement
Figures

Similar articles
-
Effects of AT1- and beta-adrenergic receptor antagonists on TGF-beta1-induced fibrosis in transgenic mice.Eur J Clin Invest. 2009 Oct;39(10):851-9. doi: 10.1111/j.1365-2362.2009.02183.x. Epub 2009 Jun 11. Eur J Clin Invest. 2009. PMID: 19522835
-
Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1).Am J Physiol Heart Circ Physiol. 2002 Sep;283(3):H1253-62. doi: 10.1152/ajpheart.00578.2001. Am J Physiol Heart Circ Physiol. 2002. PMID: 12181157
-
Inhibition of AP-1 signaling by JDP2 overexpression protects cardiomyocytes against hypertrophy and apoptosis induction.Cardiovasc Res. 2013 Jul 1;99(1):121-8. doi: 10.1093/cvr/cvt094. Epub 2013 Apr 23. Cardiovasc Res. 2013. PMID: 23612584
-
Cardiac hypertrophy induced by sustained beta-adrenoreceptor activation: pathophysiological aspects.Heart Fail Rev. 2007 Mar;12(1):66-86. doi: 10.1007/s10741-007-9007-4. Epub 2007 Mar 27. Heart Fail Rev. 2007. PMID: 17387610 Review.
-
Transforming growth factor (TGF)-β signaling in cardiac remodeling.J Mol Cell Cardiol. 2011 Oct;51(4):600-6. doi: 10.1016/j.yjmcc.2010.10.033. Epub 2010 Nov 6. J Mol Cell Cardiol. 2011. PMID: 21059352 Free PMC article. Review.
Cited by
-
Communication Between Cardiomyocytes and Fibroblasts During Cardiac Ischemia/Reperfusion and Remodeling: Roles of TGF-β, CTGF, the Renin Angiotensin Axis, and Non-coding RNA Molecules.Front Physiol. 2021 Sep 3;12:716721. doi: 10.3389/fphys.2021.716721. eCollection 2021. Front Physiol. 2021. PMID: 34539441 Free PMC article. Review.
-
Parathyroid Hormone: A Uremic Toxin.Toxins (Basel). 2020 Mar 17;12(3):189. doi: 10.3390/toxins12030189. Toxins (Basel). 2020. PMID: 32192220 Free PMC article. Review.
-
Differentiation of cardiomyocytes from amniotic fluid‑derived mesenchymal stem cells by combined induction with transforming growth factor β1 and 5‑azacytidine.Mol Med Rep. 2017 Nov;16(5):5887-5893. doi: 10.3892/mmr.2017.7373. Epub 2017 Aug 28. Mol Med Rep. 2017. PMID: 28849231 Free PMC article.
-
Molecular switches under TGFβ signalling during progression from cardiac hypertrophy to heart failure.Br J Pharmacol. 2016 Jan;173(1):3-14. doi: 10.1111/bph.13344. Epub 2015 Nov 16. Br J Pharmacol. 2016. PMID: 26431212 Free PMC article. Review.
-
Serum level of transforming growth factor beta 1 is associated with left atrial voltage in patients with chronic atrial fibrillation.Indian Pacing Electrophysiol J. 2018 May-Jun;18(3):95-99. doi: 10.1016/j.ipej.2017.11.001. Epub 2017 Nov 15. Indian Pacing Electrophysiol J. 2018. PMID: 29155027 Free PMC article.
References
-
- Massagué J, Blain SW, Lo RS. TGF-β signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. - PubMed
-
- Shi Y, Massagué J. Mechanisms of TGF-β signaling from the cell membrane to the nucleus. Cell. 2003;113:685–700. - PubMed
-
- Millan FA, Denhez F, Kondaiah P, Akhurst RJ. Embryonic gene expression patterns of TGF beta 1, beta 2, and beta 3 suggest different developmental functions in vivo. Development. 1991;111:131–143. - PubMed
-
- Brand T, Schneider MD. The TGF-β superfamily in myocardium: Ligands, receptors, transduction, and function. J Mol Cell Cardiol. 1995;27:5–18. - PubMed
-
- Rosenkranz S. TGF-β1 and angiotensin networking in cardiac remodeling. Cardiovasc Res. 2004;63:423–432. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
