

Specific capture and whole-genome sequencing of viruses from clinical samples
- PMID: 22125625
- PMCID: PMC3220689
- DOI: 10.1371/journal.pone.0027805
Specific capture and whole-genome sequencing of viruses from clinical samples
Erratum in
- PLoS One. 2012;7(1). doi:10.1371/annotation/3f1444bc-ab9d-4112-958a-2e068792f26f
Abstract
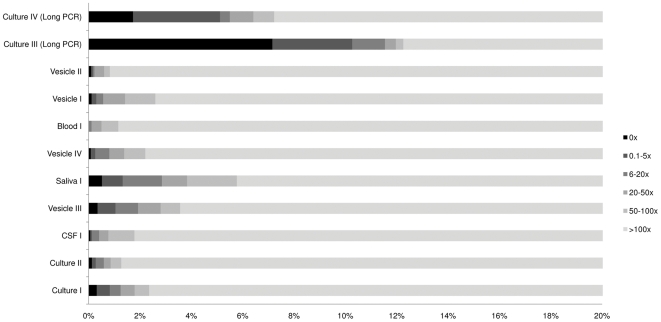
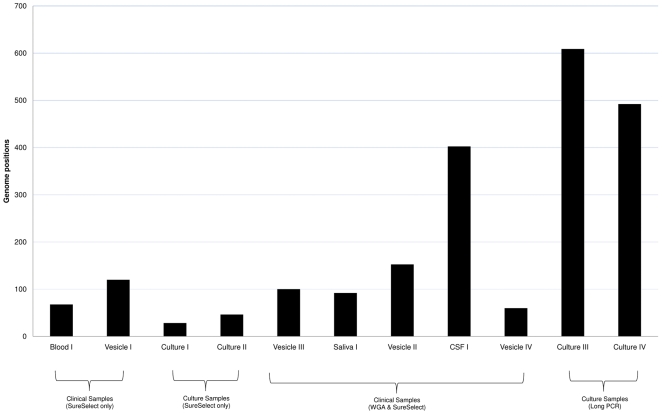
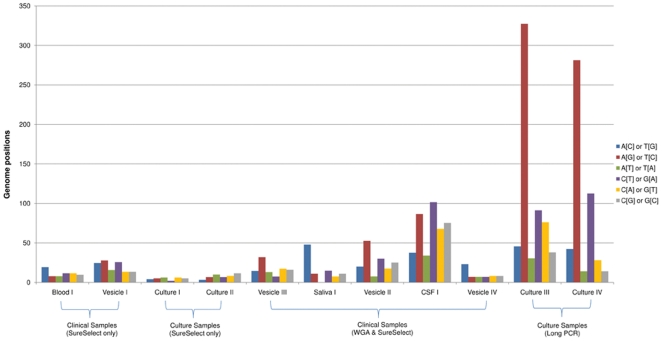
Whole genome sequencing of viruses directly from clinical samples is integral for understanding the genetics of host-virus interactions. Here, we report the use of sample sparing target enrichment (by hybridisation) for viral nucleic acid separation and deep-sequencing of herpesvirus genomes directly from a range of clinical samples including saliva, blood, virus vesicles, cerebrospinal fluid, and tumour cell lines. We demonstrate the effectiveness of the method by deep-sequencing 13 highly cell-associated human herpesvirus genomes and generating full length genome alignments at high read depth. Moreover, we show the specificity of the method enables the study of viral population structures and their diversity within a range of clinical samples types.
Conflict of interest statement
Figures
References
-
- Kew O, Morris-Glasgow V, Landaverde M, Burns C, Shaw J, et al. Outbreak of poliomyelitis in Hispaniola associated with circulating type 1 vaccine-derived poliovirus. Science. 2002;296:356–359. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
