

Statistical analysis of rare sequence variants: an overview of collapsing methods
- PMID: 22128052
- PMCID: PMC3277891
- DOI: 10.1002/gepi.20643
Statistical analysis of rare sequence variants: an overview of collapsing methods
Abstract
With the advent of novel sequencing technologies, interest in the identification of rare variants that influence common traits has increased rapidly. Standard statistical methods, such as the Cochrane-Armitage trend test or logistic regression, fail in this setting for the analysis of unrelated subjects because of the rareness of the variants. Recently, various alternative approaches have been proposed that circumvent the rareness problem by collapsing rare variants in a defined genetic region or sets of regions. We provide an overview of these collapsing methods for association analysis and discuss the use of permutation approaches for significance testing of the data-adaptive methods.
© 2011 Wiley Periodicals, Inc.
Figures
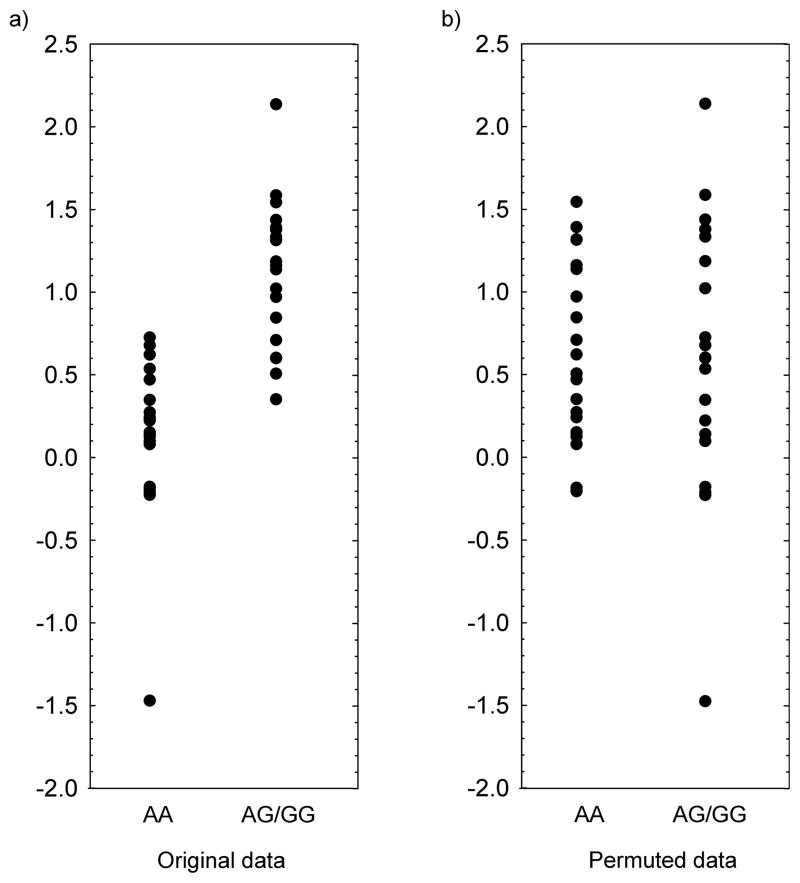
References
-
- Asimit J, Zeggini E. Rare variant association analysis methods for complex traits. Annu Rev Genet. 2010;44:293–308. - PubMed
-
- Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, Tavtigian SV, Monteiro AN, Iversen ES, Couch FJ, et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81(5):873–83. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
