

An overview of the c-MET signaling pathway
- PMID: 22128289
- PMCID: PMC3225017
- DOI: 10.1177/1758834011422556
An overview of the c-MET signaling pathway
Abstract
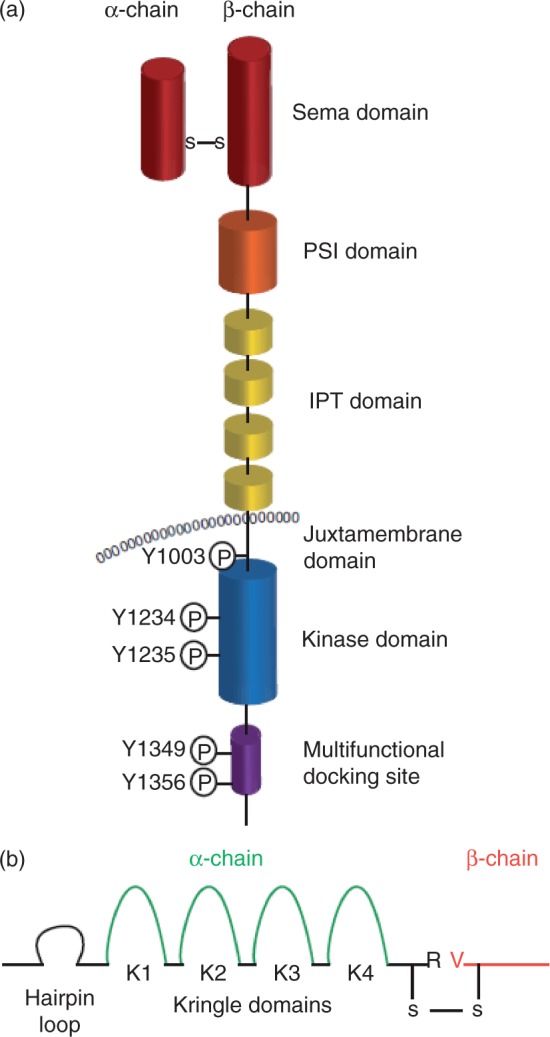
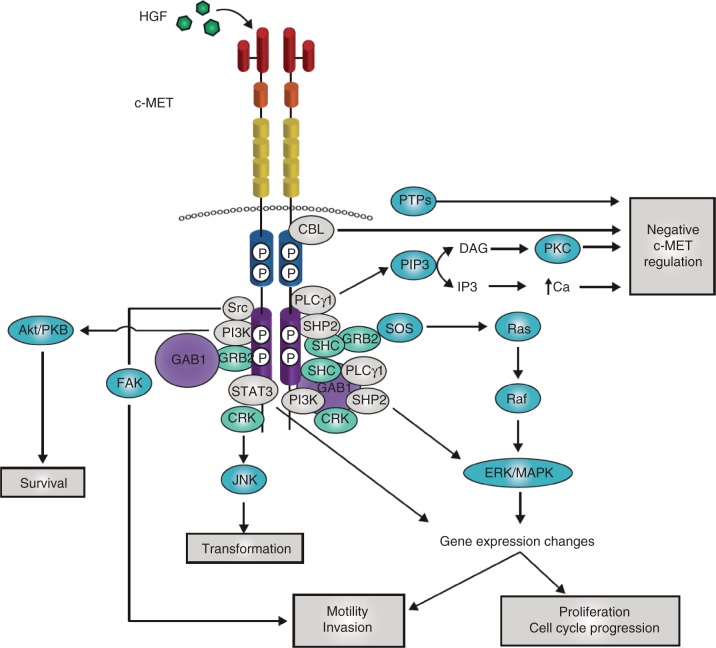
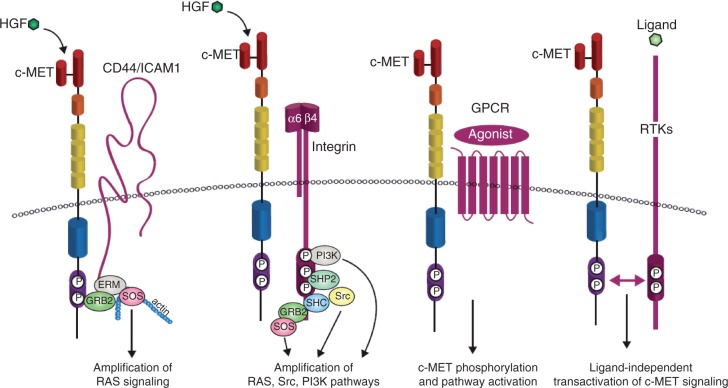
c-MET is a receptor tyrosine kinase that, after binding with its ligand, hepatocyte growth factor, activates a wide range of different cellular signaling pathways, including those involved in proliferation, motility, migration and invasion. Although c-MET is important in the control of tissue homeostasis under normal physiological conditions, it has also been found to be aberrantly activated in human cancers via mutation, amplification or protein overexpression. This paper provides an overview of the c-MET signaling pathway, including its role in the development of cancers, and provides a rationale for targeting the pathway as a possible treatment option.
Keywords: MET; c-MET; cancer; hepatocyte growth factor (HGF); receptor tyrosine kinase; signaling.
Figures
References
-
- Abounader R., Reznik T., Colantuoni C., Martinez-Murillo F., Rosen E.M., Laterra J. (2004) Regulation of c-Met-dependent gene expression by PTEN. Oncogene 23: 9173–9182 - PubMed
-
- Bachleitner-Hofmann T., Sun M.Y., Chen C.T., Tang L., Song L., Zeng Z., et al. (2008) HER kinase activation confers resistance to MET tyrosine kinase inhibition in MET oncogene-addicted gastric cancer cells. Mol Cancer Ther 7: 3499–3508 - PubMed
-
- Benvenuti S., Lazzari L., Arnesano A., Li Chiavi G., Gentile A., Comoglio P.M. (2011) Ron kinase transphosphorylation sustains MET oncogene addiction. Cancer Res 71: 1945–1955 - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
