

Human herpesvirus 8 viral interleukin-6 interacts with splice variant 2 of vitamin K epoxide reductase complex subunit 1
- PMID: 22130532
- PMCID: PMC3264350
- DOI: 10.1128/JVI.05782-11
Human herpesvirus 8 viral interleukin-6 interacts with splice variant 2 of vitamin K epoxide reductase complex subunit 1
Abstract
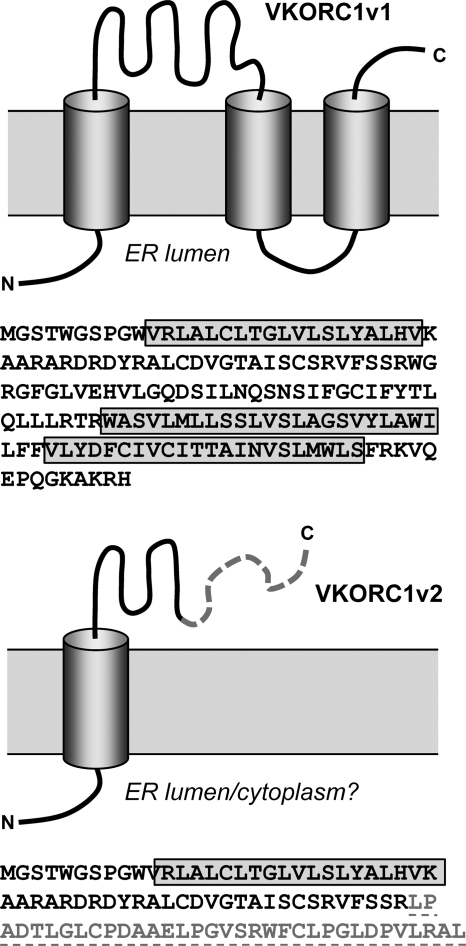
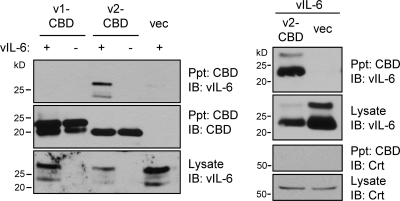
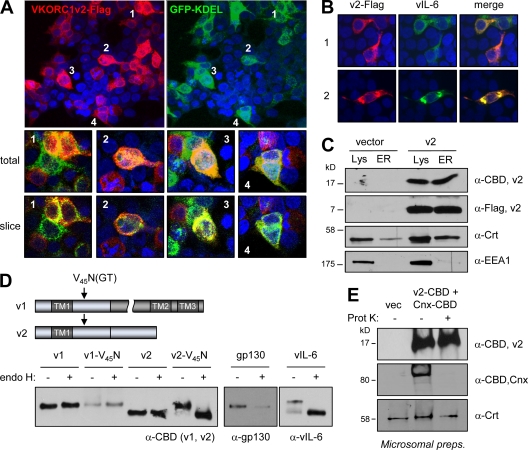
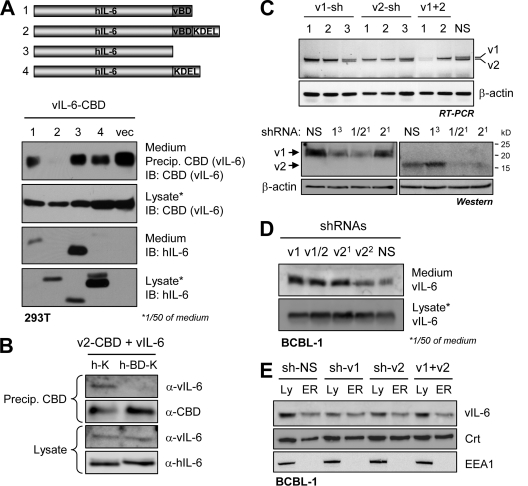
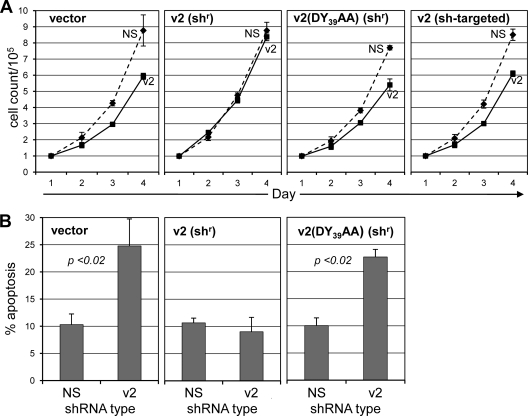
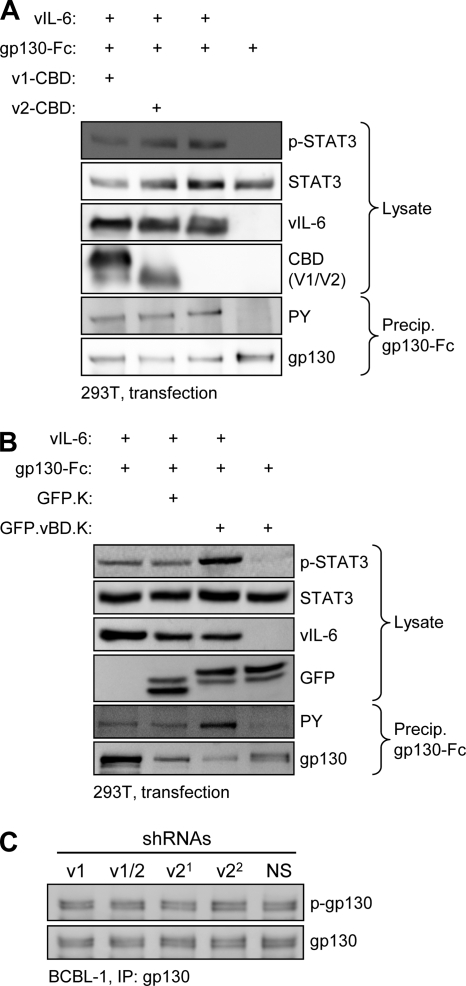
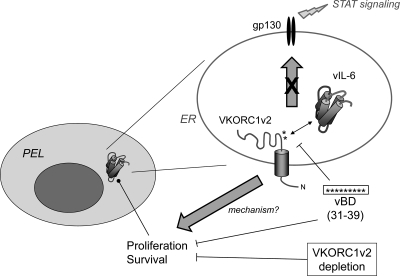
Viral interleukin-6 (vIL-6) specified by human herpesvirus 8 is, unlike its cellular counterpart, secreted very inefficiently and can signal via vIL-6(2):gp130(2) signaling complexes from the endoplasmic reticulum (ER) compartment. Intracellular, autocrine activities of vIL-6 are important for proproliferative and prosurvival activities of the viral cytokine in latently infected primary effusion lymphoma (PEL) cells. However, the molecular determinants of vIL-6 ER localization and function are unclear. Using yeast two-hybrid analysis, we identified the database-documented but uncharacterized splice variant of vitamin K epoxide reductase complex subunit 1 (VKORC1), termed VKORC1 variant 2 (VKORC1v2), as a potential interaction partner of vIL-6. In transfected cells, epitope-tagged VKORC1v2 was found to localize to the ER, to adopt a single-transmembrane (TM) topology placing the C tail in the ER lumen, and to bind vIL-6 via these sequences. Deletion mutagenesis and coprecipitation assays mapped the vIL-6-binding domain (vBD) of VKORC1v2 to TM-proximal residues 31 to 39. However, while sufficient to confer vIL-6 binding to a heterologous protein, vBD was unable to induce vIL-6 secretion when fused to (secreted) hIL-6, suggesting a VKORC1v2-independent mechanism of vIL-6 ER retention. In functional assays, overexpression of ER-directed vBD led to suppression of PEL cell proliferation and viability, effects also mediated by VKORC1v2 depletion and, as reported previously, by vIL-6 suppression. The growth-inhibitory and proapoptotic effects of VKORC1v2 depletion could be rescued by transduced wild-type VKORC1v2 but not by a vIL-6-refractory vBD-altered variant, indicating the functional relevance of the vIL-6-VKORC1v2 interaction. Notably, gp130 signaling was unaffected by VKORC1v2 or vBD overexpression or by VKORC1v2 depletion, suggesting an alternative pathway of vIL-6 activity via VKORC1v2. Combined, our data identify a novel and functionally significant interaction partner of vIL-6 that could potentially be targeted for therapeutic benefit.
Figures


References
-
- Cain D, Hutson SM, Wallin R. 1997. Assembly of the warfarin-sensitive vitamin K 2,3-epoxide reductase enzyme complex in the endoplasmic reticulum membrane. J. Biol. Chem. 272:29068–29075 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
