

Cyclic AMP-dependent protein kinase A regulates the alternative splicing of CaMKIIδ
- PMID: 22132070
- PMCID: PMC3222655
- DOI: 10.1371/journal.pone.0025745
Cyclic AMP-dependent protein kinase A regulates the alternative splicing of CaMKIIδ
Abstract
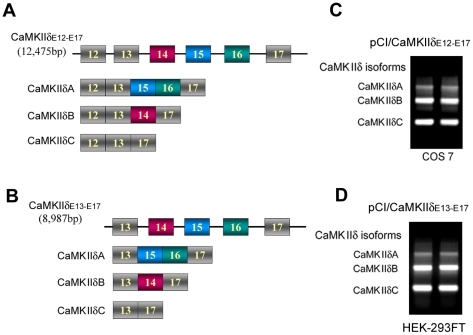
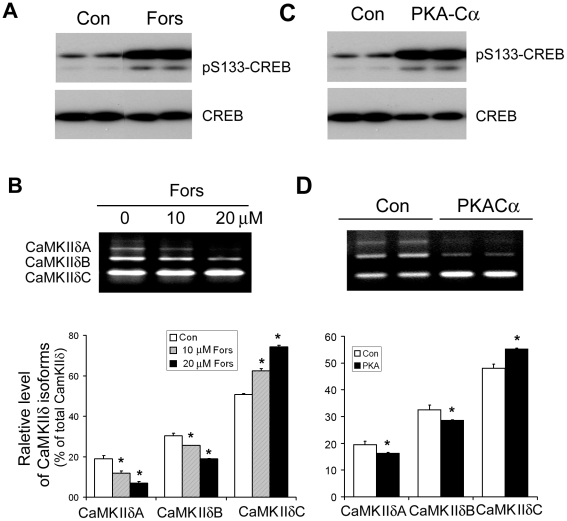
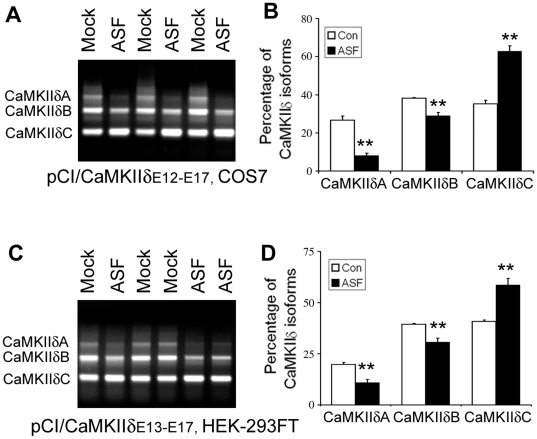
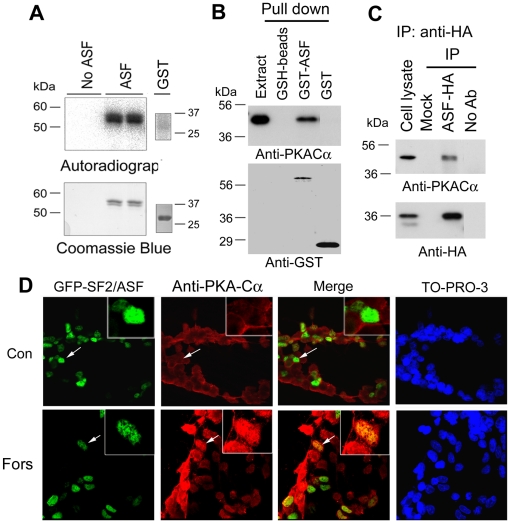
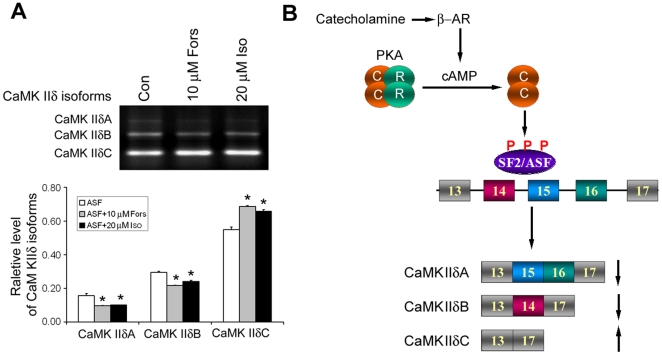
Ca(2+)/calmodulin-dependent protein kinase (CaMK) IIδ is predominantly expressed in the heart. There are three isoforms of CaMKIIδ resulting from the alternative splicing of exons 14, 15, and 16 of its pre-mRNA, which is regulated by the splicing factor SF2/ASF. Inclusion of exons 15 and 16 or of exon 14 generates δA or δB isoform. The exclusion of all three exons gives rise to δC isoform, which is selectively increased in pressure-overload-induced hypertrophy. Overexpression of either δB or δC induces hypertrophy and heart failure, suggesting their specific role in the pathogenesis of hypertrophy and heart failure. It is well known that the β-adrenergic-cyclic AMP-dependent protein kinase A (PKA) pathway is implicated in heart failure. To determine the role of PKA in the alternative splicing of CaMKIIδ, we constructed mini-CaMKIIδ genes and used these genes to investigate the regulation of the alternative splicing of CaMKIIδ by PKA in cultured cells. We found that PKA promoted the exclusion of exons 14, 15, and 16 of CaMKIIδ, resulting in an increase in δC isoform. PKA interacted with and phosphorylated SF2/ASF, and enhanced SF2/ASF's activity to promote the exclusion of exons 14, 15, and 16 of CaMKIIδ, leading to a further increase in the expression of δC isoform. These findings suggest that abnormality in β-adrenergic-PKA signaling may contribute to cardiomyopathy and heart failure through dysregulation in the alternative splicing of CaMKIIδ exons 14, 15, and 16 and up-regulation of CaMKIIδC.
Conflict of interest statement
Figures
Similar articles
-
ASF/SF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle.Cell. 2005 Jan 14;120(1):59-72. doi: 10.1016/j.cell.2004.11.036. Cell. 2005. PMID: 15652482
-
Involvement of the dual-specificity tyrosine phosphorylation-regulated kinase 1A-alternative splicing factor-calcium/calmodulin-dependent protein kinase IIδ signaling pathway in myocardial infarction-induced heart failure of rats.J Card Fail. 2015 Sep;21(9):751-60. doi: 10.1016/j.cardfail.2015.05.015. Epub 2015 Jun 9. J Card Fail. 2015. PMID: 26067684
-
Cyclic AMP-dependent protein kinase regulates the alternative splicing of tau exon 10: a mechanism involved in tau pathology of Alzheimer disease.J Biol Chem. 2011 Apr 22;286(16):14639-48. doi: 10.1074/jbc.M110.204453. Epub 2011 Mar 2. J Biol Chem. 2011. PMID: 21367856 Free PMC article.
-
Role of Ca2+/calmodulin-dependent protein kinase II in cardiac hypertrophy and heart failure.Cardiovasc Res. 2004 Aug 15;63(3):476-86. doi: 10.1016/j.cardiores.2004.04.026. Cardiovasc Res. 2004. PMID: 15276473 Review.
-
Cardiomyocyte calcium and calcium/calmodulin-dependent protein kinase II: friends or foes?Recent Prog Horm Res. 2004;59:141-68. doi: 10.1210/rp.59.1.141. Recent Prog Horm Res. 2004. PMID: 14749501 Review.
Cited by
-
CaMKIIdelta subtypes: localization and function.Front Pharmacol. 2014 Feb 11;5:15. doi: 10.3389/fphar.2014.00015. eCollection 2014. Front Pharmacol. 2014. PMID: 24575042 Free PMC article. Review.
-
The Molecular Basis for Specificity at the Level of the Protein Kinase a Catalytic Subunit.Front Endocrinol (Lausanne). 2018 Sep 12;9:538. doi: 10.3389/fendo.2018.00538. eCollection 2018. Front Endocrinol (Lausanne). 2018. PMID: 30258407 Free PMC article. Review.
-
Post-Transcriptional Modification by Alternative Splicing and Pathogenic Splicing Variants in Cardiovascular Development and Congenital Heart Defects.Int J Mol Sci. 2023 Jan 13;24(2):1555. doi: 10.3390/ijms24021555. Int J Mol Sci. 2023. PMID: 36675070 Free PMC article. Review.
-
Genetically Encoded Fluorescent Biosensors Illuminate the Spatiotemporal Regulation of Signaling Networks.Chem Rev. 2018 Dec 26;118(24):11707-11794. doi: 10.1021/acs.chemrev.8b00333. Epub 2018 Dec 14. Chem Rev. 2018. PMID: 30550275 Free PMC article. Review.
-
Alternative splicing isoforms in health and disease.Pflugers Arch. 2018 Jul;470(7):995-1016. doi: 10.1007/s00424-018-2136-x. Epub 2018 Mar 13. Pflugers Arch. 2018. PMID: 29536164 Review.
References
-
- Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, et al. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–1293. - PubMed
-
- Zhang Q, Li JX, Zheng JW, Liu RK, Liang JH. L-type Ca(2+) channel blockers inhibit the development but not the expression of sensitization to morphine in mice. Eur J Pharmacol. 2003;467:145–150. - PubMed
-
- Zhang T, Kohlhaas M, Backs J, Mishra S, Phillips W, et al. CaMKIIdelta isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J Biol Chem. 2007;282:35078–35087. - PubMed
-
- Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, et al. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912–919. - PubMed
-
- Zhang ZS, Cheng HJ, Onishi K, Ohte N, Wannenburg T, et al. Enhanced inhibition of L-type Ca2+ current by beta3-adrenergic stimulation in failing rat heart. J Pharmacol Exp Ther. 2005;315:1203–1211. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
