

A novel mutation in the HSD17B10 gene of a 10-year-old boy with refractory epilepsy, choreoathetosis and learning disability
- PMID: 22132097
- PMCID: PMC3222643
- DOI: 10.1371/journal.pone.0027348
A novel mutation in the HSD17B10 gene of a 10-year-old boy with refractory epilepsy, choreoathetosis and learning disability
Abstract
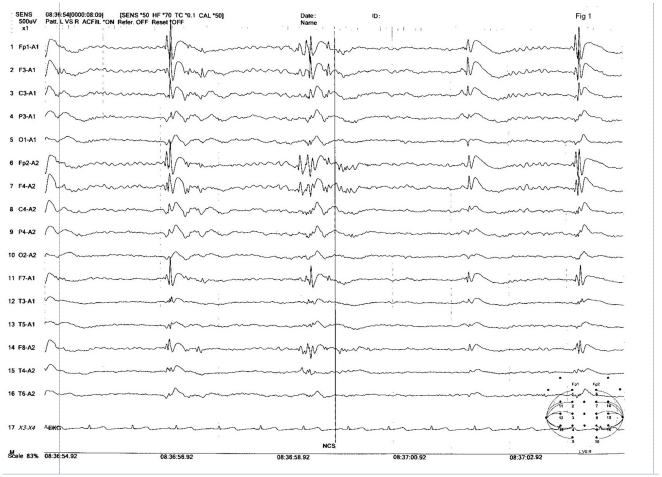
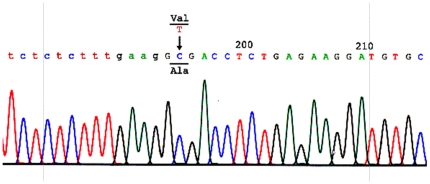
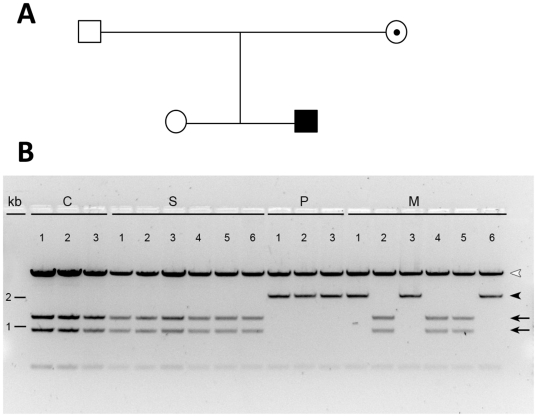
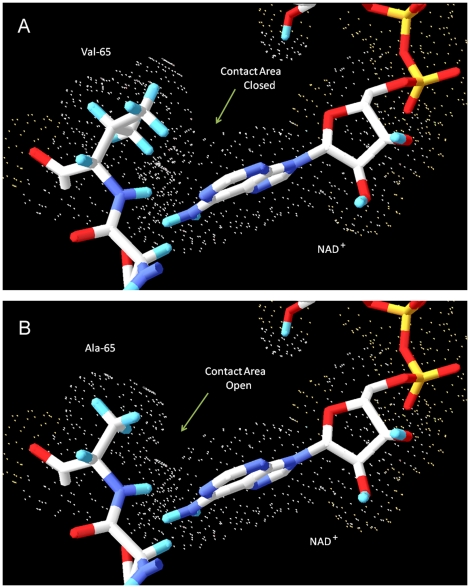
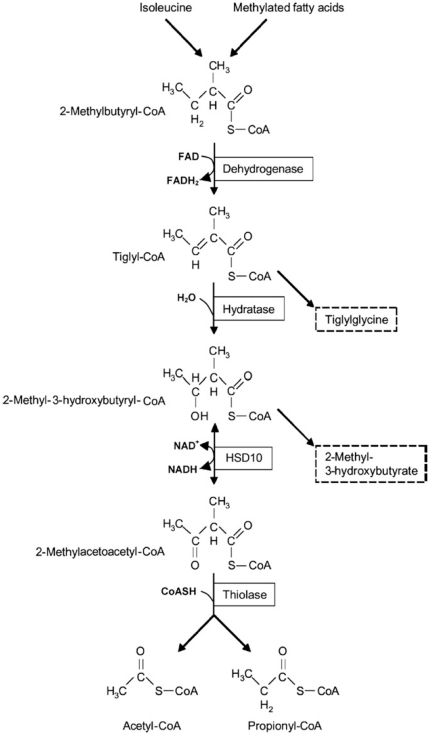
Hydroxysteroid (17beta) dehydrogenase 10 (HSD10) is a mitochondrial multifunctional enzyme encoded by the HSD17B10 gene. Missense mutations in this gene result in HSD10 deficiency, whereas a silent mutation results in mental retardation, X-linked, syndromic 10 (MRXS10). Here we report a novel missense mutation found in the HSD17B10 gene, namely c.194T>C transition (rs104886492), brought about by the loss of two forked methyl groups of valine 65 in the HSD10 active site. The affected boy, who possesses mutant HSD10 (p.V65A), has a neurological syndrome with metabolic derangements, choreoathetosis, refractory epilepsy and learning disability. He has no history of acute decompensation or metabolic acidosis whereas his urine organic acid profile, showing elevated levels of 2-methyl-3-hydroxybutyrate and tiglylglycine, is characteristic of HSD10 deficiency. His HSD10 activity was much lower than the normal control level, with normal β-ketothiolase activity. The c.194T>C mutation in HSD17B10 can be identified by the restriction fragment polymorphism analysis, thereby facilitating the screening of this novel mutation in individuals with intellectual disability of unknown etiology and their family members much easier. The patient's mother is an asymptomatic carrier, and has a mixed ancestry (Hawaiian, Japanese and Chinese). This demonstrates that HSD10 deficiency patients are not confined to a particular ethnicity although previously reported cases were either Spanish or German descendants.
Conflict of interest statement
Figures

Similar articles
-
Clinical and molecular analysis of 6 Chinese patients with isoleucine metabolism defects: identification of 3 novel mutations in the HSD17B10 and ACAT1 gene.Metab Brain Dis. 2017 Dec;32(6):2063-2071. doi: 10.1007/s11011-017-0097-y. Epub 2017 Sep 5. Metab Brain Dis. 2017. PMID: 28875337
-
The first case in Asia of 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency (HSD10 disease) with atypical presentation.J Hum Genet. 2014 Nov;59(11):609-14. doi: 10.1038/jhg.2014.79. Epub 2014 Sep 18. J Hum Genet. 2014. PMID: 25231369
-
Hydroxysteroid (17β) dehydrogenase X in human health and disease.Mol Cell Endocrinol. 2011 Aug 22;343(1-2):1-6. doi: 10.1016/j.mce.2011.06.011. Epub 2011 Jun 25. Mol Cell Endocrinol. 2011. PMID: 21708223 Review.
-
A novel c.59 C > T variant of the HSD17B10 gene as a possible cause of the neonatal form of HSD10 mitochondrial disease with hepatic dysfunction: a case report and review of the literature.Orphanet J Rare Dis. 2025 Mar 7;20(1):108. doi: 10.1186/s13023-024-03513-2. Orphanet J Rare Dis. 2025. PMID: 40055822 Free PMC article. Review.
-
Mental retardation linked to mutations in the HSD17B10 gene interfering with neurosteroid and isoleucine metabolism.Proc Natl Acad Sci U S A. 2009 Sep 1;106(35):14820-4. doi: 10.1073/pnas.0902377106. Epub 2009 Aug 17. Proc Natl Acad Sci U S A. 2009. PMID: 19706438 Free PMC article.
Cited by
-
Infantile Neurodegeneration Results from Mutants of 17β-Hydroxysteroid Dehydrogenase Type 10 Rather Than Aβ-Binding Alcohol Dehydrogenase.Int J Mol Sci. 2023 May 9;24(10):8487. doi: 10.3390/ijms24108487. Int J Mol Sci. 2023. PMID: 37239833 Free PMC article. Review.
-
Involvement of Type 10 17β-Hydroxysteroid Dehydrogenase in the Pathogenesis of Infantile Neurodegeneration and Alzheimer's Disease.Int J Mol Sci. 2023 Dec 18;24(24):17604. doi: 10.3390/ijms242417604. Int J Mol Sci. 2023. PMID: 38139430 Free PMC article. Review.
-
3-Hydroxyacyl-CoA and Alcohol Dehydrogenase Activities of Mitochondrial Type 10 17β-Hydroxysteroid Dehydrogenase in Neurodegeneration Study.J Alzheimers Dis. 2022;88(4):1487-1497. doi: 10.3233/JAD-220481. J Alzheimers Dis. 2022. PMID: 35786658 Free PMC article.
-
Clinical and molecular analysis of 6 Chinese patients with isoleucine metabolism defects: identification of 3 novel mutations in the HSD17B10 and ACAT1 gene.Metab Brain Dis. 2017 Dec;32(6):2063-2071. doi: 10.1007/s11011-017-0097-y. Epub 2017 Sep 5. Metab Brain Dis. 2017. PMID: 28875337
-
Roles of Type 10 17β-Hydroxysteroid Dehydrogenase in Health and Disease.J Pers Med. 2025 Aug 1;15(8):346. doi: 10.3390/jpm15080346. J Pers Med. 2025. PMID: 40863408 Free PMC article. Review.
References
-
- Yang SY, He XY, Schulz H. Multiple functions of type 10 17beta-hydroxysteroid dehydrogenase. Trends Endocrinol Metab. 2005;16:167–175. - PubMed
-
- Yang SY, He XY, Miller D. HSD17B10: A gene involved in cognitive function through metabolism of isoleucine and neuroactive steroids. Mol Genet Metab. 2007;92:36–42. - PubMed
-
- He XY, Merz G, Mehta P, Schulz H, Yang SY. Human brain short chain L-3-hydroxyacyl coenzyme A dehydrogenase is a single-domain multifunctional enzyme. Characterization of a novel 17β-hydroxysteroid dehydrogenase. J Biol Chem. 1999;274:15014–15019. - PubMed
-
- He XY, Merz G, Yang YZ, Schulz H, Yang SY. Characterization and localization of human type 10 17β-hydroxysteroid dehydrogenase. Eur J Biochem. 2001;268:4899–4907. - PubMed
-
- He XY, Wegiel J, Yang SY. Intracellular oxidation of allopregnanolone by human brain type 10 17beta-hydroxysteroid dehydrogenase. Brain Res. 2005;1040:29–35. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
