

Modeling of human prokineticin receptors: interactions with novel small-molecule binders and potential off-target drugs
- PMID: 22132188
- PMCID: PMC3221691
- DOI: 10.1371/journal.pone.0027990
Modeling of human prokineticin receptors: interactions with novel small-molecule binders and potential off-target drugs
Abstract
Background and motivation: The Prokineticin receptor (PKR) 1 and 2 subtypes are novel members of family A GPCRs, which exhibit an unusually high degree of sequence similarity. Prokineticins (PKs), their cognate ligands, are small secreted proteins of ∼80 amino acids; however, non-peptidic low-molecular weight antagonists have also been identified. PKs and their receptors play important roles under various physiological conditions such as maintaining circadian rhythm and pain perception, as well as regulating angiogenesis and modulating immunity. Identifying binding sites for known antagonists and for additional potential binders will facilitate studying and regulating these novel receptors. Blocking PKRs may serve as a therapeutic tool for various diseases, including acute pain, inflammation and cancer.
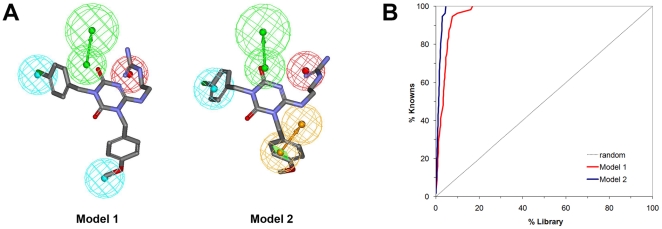
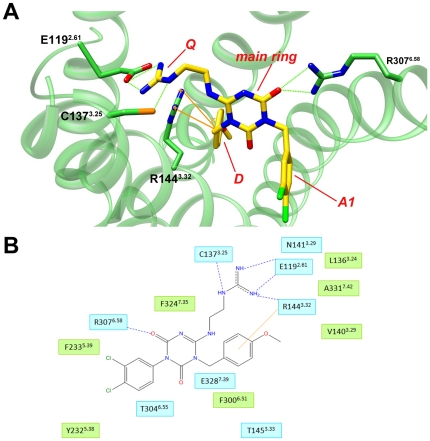
Methods and results: Ligand-based pharmacophore models were derived from known antagonists, and virtual screening performed on the DrugBank dataset identified potential human PKR (hPKR) ligands with novel scaffolds. Interestingly, these included several HIV protease inhibitors for which endothelial cell dysfunction is a documented side effect. Our results suggest that the side effects might be due to inhibition of the PKR signaling pathway. Docking of known binders to a 3D homology model of hPKR1 is in agreement with the well-established canonical TM-bundle binding site of family A GPCRs. Furthermore, the docking results highlight residues that may form specific contacts with the ligands. These contacts provide structural explanation for the importance of several chemical features that were obtained from the structure-activity analysis of known binders. With the exception of a single loop residue that might be perused in the future for obtaining subtype-specific regulation, the results suggest an identical TM-bundle binding site for hPKR1 and hPKR2. In addition, analysis of the intracellular regions highlights variable regions that may provide subtype specificity.
Conflict of interest statement
Figures







Similar articles
-
The different ligand-binding modes of relaxin family peptide receptors RXFP1 and RXFP2.Mol Endocrinol. 2012 Nov;26(11):1896-906. doi: 10.1210/me.2012-1188. Epub 2012 Sep 12. Mol Endocrinol. 2012. PMID: 22973049 Free PMC article.
-
Homology model-assisted elucidation of binding sites in GPCRs.Methods Mol Biol. 2012;914:179-205. doi: 10.1007/978-1-62703-023-6_11. Methods Mol Biol. 2012. PMID: 22976029 Review.
-
Recent advances in structure-based virtual screening of G-protein coupled receptors.AAPS J. 2009 Mar;11(1):178-85. doi: 10.1208/s12248-009-9094-3. Epub 2009 Mar 17. AAPS J. 2009. PMID: 19291412 Free PMC article. Review.
-
Exploration of the structural requirements of HIV-protease inhibitors using pharmacophore, virtual screening and molecular docking approaches for lead identification.J Mol Graph Model. 2015 Mar;56:20-30. doi: 10.1016/j.jmgm.2014.11.015. Epub 2014 Dec 5. J Mol Graph Model. 2015. PMID: 25541527
-
First pharmacophore model of CCR3 receptor antagonists and its homology model-assisted, stepwise virtual screening.Chem Biol Drug Des. 2011 May;77(5):373-87. doi: 10.1111/j.1747-0285.2011.01088.x. Epub 2011 Mar 1. Chem Biol Drug Des. 2011. PMID: 21284830
Cited by
-
Structure-activity relationships of FMRF-NH2 peptides demonstrate A role for the conserved C terminus and unique N-terminal extension in modulating cardiac contractility.PLoS One. 2013 Sep 17;8(9):e75502. doi: 10.1371/journal.pone.0075502. eCollection 2013. PLoS One. 2013. PMID: 24069424 Free PMC article.
-
Design and synthesis of novel protein kinase R (PKR) inhibitors.Mol Divers. 2016 Nov;20(4):805-819. doi: 10.1007/s11030-016-9689-4. Epub 2016 Aug 1. Mol Divers. 2016. PMID: 27480630
-
Toll-like receptor 9 interaction with CpG ODN--an in silico analysis approach.Theor Biol Med Model. 2013 Mar 14;10:18. doi: 10.1186/1742-4682-10-18. Theor Biol Med Model. 2013. PMID: 23497207 Free PMC article.
-
Discovery and cardioprotective effects of the first non-Peptide agonists of the G protein-coupled prokineticin receptor-1.PLoS One. 2015 Apr 1;10(4):e0121027. doi: 10.1371/journal.pone.0121027. eCollection 2015. PLoS One. 2015. PMID: 25831128 Free PMC article.
-
A Novel Drug-Mouse Phenotypic Similarity Method Detects Molecular Determinants of Drug Effects.PLoS Comput Biol. 2016 Sep 27;12(9):e1005111. doi: 10.1371/journal.pcbi.1005111. eCollection 2016 Sep. PLoS Comput Biol. 2016. PMID: 27673331 Free PMC article.
References
-
- LeCouter J, Kowalski J, Foster J, Hass P, Zhang Z, et al. Identification of an angiogenic mitogen selective for endocrine gland endothelium. Nature. 2001;412:877–884. - PubMed
-
- Li M, Bullock CM, Knauer DJ, Ehlert FJ, Zhou QY. Identification of two prokineticin cDNAs: recombinant proteins potently contract gastrointestinal smooth muscle. Mol Pharmacol. 2001;59:692–698. - PubMed
-
- Masuda Y, Takatsu Y, Terao Y, Kumano S, Ishibashi Y, et al. Isolation and identification of EG-VEGF/prokineticins as cognate ligands for two orphan G-protein-coupled receptors. Biochem Biophys Res Commun. 2002;293:396–402. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
