

Relative impact of key sources of systematic noise in Affymetrix and Illumina gene-expression microarray experiments
- PMID: 22133085
- PMCID: PMC3269440
- DOI: 10.1186/1471-2164-12-589
Relative impact of key sources of systematic noise in Affymetrix and Illumina gene-expression microarray experiments
Abstract
Background: Systematic processing noise, which includes batch effects, is very common in microarray experiments but is often ignored despite its potential to confound or compromise experimental results. Compromised results are most likely when re-analysing or integrating datasets from public repositories due to the different conditions under which each dataset is generated. To better understand the relative noise-contributions of various factors in experimental-design, we assessed several Illumina and Affymetrix datasets for technical variation between replicate hybridisations of Universal Human Reference (UHRR) and individual or pooled breast-tumour RNA.
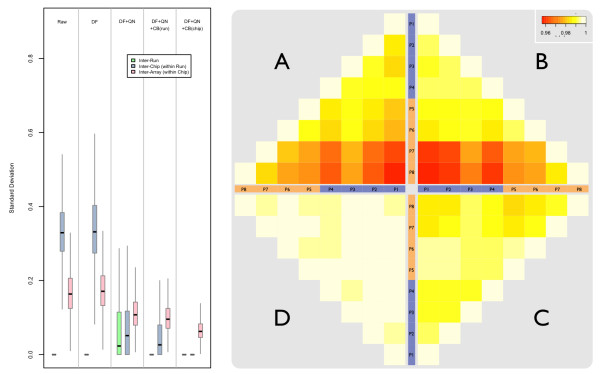
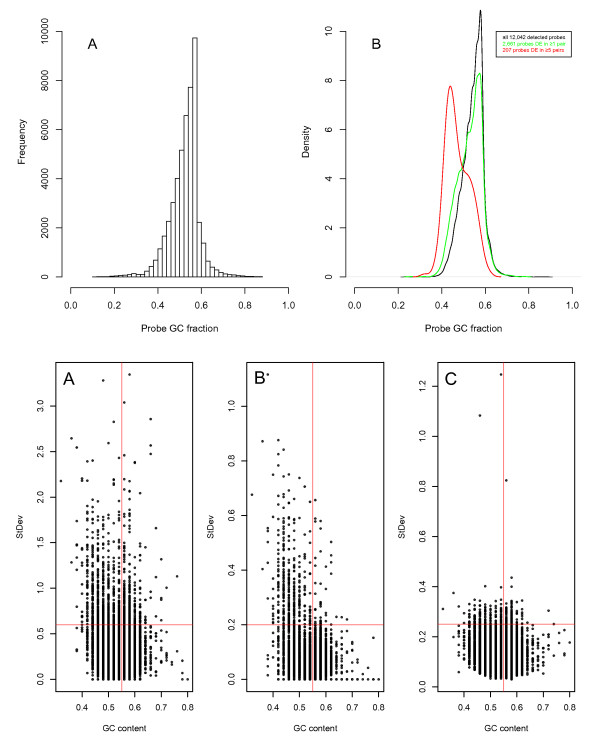
Results: A varying degree of systematic noise was observed in each of the datasets, however in all cases the relative amount of variation between standard control RNA replicates was found to be greatest at earlier points in the sample-preparation workflow. For example, 40.6% of the total variation in reported expressions were attributed to replicate extractions, compared to 13.9% due to amplification/labelling and 10.8% between replicate hybridisations. Deliberate probe-wise batch-correction methods were effective in reducing the magnitude of this variation, although the level of improvement was dependent on the sources of noise included in the model. Systematic noise introduced at the chip, run, and experiment levels of a combined Illumina dataset were found to be highly dependent upon the experimental design. Both UHRR and pools of RNA, which were derived from the samples of interest, modelled technical variation well although the pools were significantly better correlated (4% average improvement) and better emulated the effects of systematic noise, over all probes, than the UHRRs. The effect of this noise was not uniform over all probes, with low GC-content probes found to be more vulnerable to batch variation than probes with a higher GC-content.
Conclusions: The magnitude of systematic processing noise in a microarray experiment is variable across probes and experiments, however it is generally the case that procedures earlier in the sample-preparation workflow are liable to introduce the most noise. Careful experimental design is important to protect against noise, detailed meta-data should always be provided, and diagnostic procedures should be routinely performed prior to downstream analyses for the detection of bias in microarray studies.
Figures
Similar articles
-
Correcting for intra-experiment variation in Illumina BeadChip data is necessary to generate robust gene-expression profiles.BMC Genomics. 2010 Feb 24;11:134. doi: 10.1186/1471-2164-11-134. BMC Genomics. 2010. PMID: 20181233 Free PMC article.
-
A revised design for microarray experiments to account for experimental noise and uncertainty of probe response.PLoS One. 2014 Mar 11;9(3):e91295. doi: 10.1371/journal.pone.0091295. eCollection 2014. PLoS One. 2014. PMID: 24618910 Free PMC article.
-
Universal Reference RNA as a standard for microarray experiments.BMC Genomics. 2004 Mar 9;5(1):20. doi: 10.1186/1471-2164-5-20. BMC Genomics. 2004. PMID: 15113400 Free PMC article.
-
Interactively optimizing signal-to-noise ratios in expression profiling: project-specific algorithm selection and detection p-value weighting in Affymetrix microarrays.Bioinformatics. 2004 Nov 1;20(16):2534-44. doi: 10.1093/bioinformatics/bth280. Epub 2004 Apr 29. Bioinformatics. 2004. PMID: 15117752
-
Comparison of gene expression microarray data with count-based RNA measurements informs microarray interpretation.BMC Genomics. 2014 Aug 4;15(1):649. doi: 10.1186/1471-2164-15-649. BMC Genomics. 2014. PMID: 25091430 Free PMC article.
Cited by
-
Methods that remove batch effects while retaining group differences may lead to exaggerated confidence in downstream analyses.Biostatistics. 2016 Jan;17(1):29-39. doi: 10.1093/biostatistics/kxv027. Epub 2015 Aug 13. Biostatistics. 2016. PMID: 26272994 Free PMC article.
-
Identification of early liver toxicity gene biomarkers using comparative supervised machine learning.Sci Rep. 2020 Nov 5;10(1):19128. doi: 10.1038/s41598-020-76129-8. Sci Rep. 2020. PMID: 33154507 Free PMC article.
-
Gene expression prediction using low-rank matrix completion.BMC Bioinformatics. 2016 Jun 17;17(1):243. doi: 10.1186/s12859-016-1106-6. BMC Bioinformatics. 2016. PMID: 27317252 Free PMC article.
-
Batch-effect detection, correction and characterisation in Illumina HumanMethylation450 and MethylationEPIC BeadChip array data.Clin Epigenetics. 2022 Apr 29;14(1):58. doi: 10.1186/s13148-022-01277-9. Clin Epigenetics. 2022. PMID: 35488315 Free PMC article.
-
Microarray Meta-Analysis and Cross-Platform Normalization: Integrative Genomics for Robust Biomarker Discovery.Microarrays (Basel). 2015 Aug 21;4(3):389-406. doi: 10.3390/microarrays4030389. Microarrays (Basel). 2015. PMID: 27600230 Free PMC article. Review.
References
-
- Kuo WP, Jenssen TK, Butte AJ, Ohno-Machado L, Kohane IS. Analysis of matched mRNA measurements from two different microarray technologies. Bioinformatics (Oxford, England) 2002. - PubMed
-
- Lander ES. Array of hope. Nature Genetics. 1999. - PubMed
-
- Sims AH. Bioinformatics and breast cancer: what can high-throughput genomic approaches actually tell us? Journal of Clinical Pathology. 2009. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
