

RNA-seq in grain unveils fate of neo- and paleopolyploidization events in bread wheat (Triticum aestivum L.)
- PMID: 22136458
- PMCID: PMC3334614
- DOI: 10.1186/gb-2011-12-12-r119
RNA-seq in grain unveils fate of neo- and paleopolyploidization events in bread wheat (Triticum aestivum L.)
Abstract
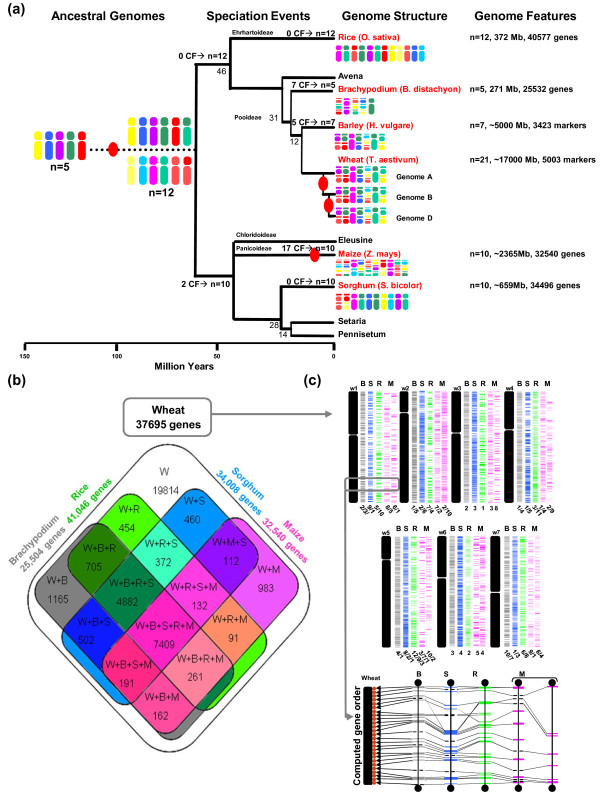
Background: Whole genome duplication is a common evolutionary event in plants. Bread wheat (Triticum aestivum L.) is a good model to investigate the impact of paleo- and neoduplications on the organization and function of modern plant genomes.
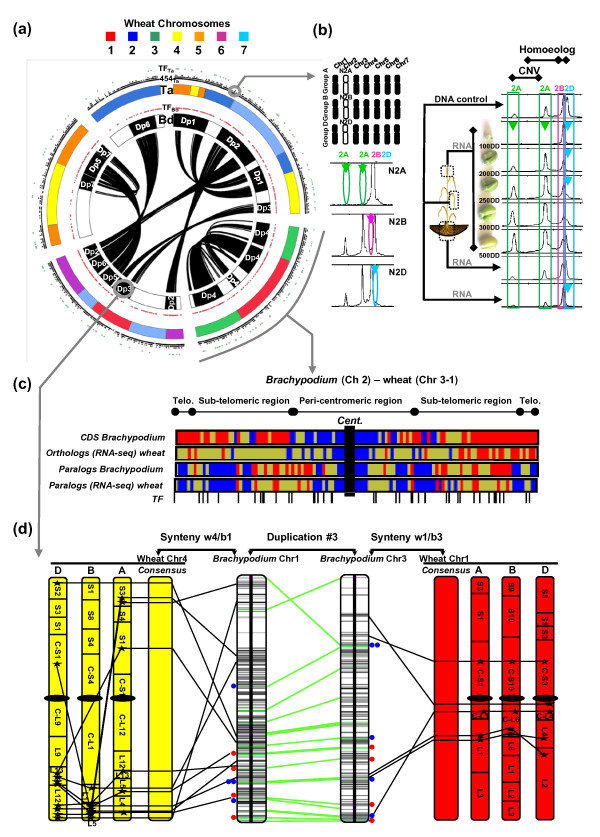
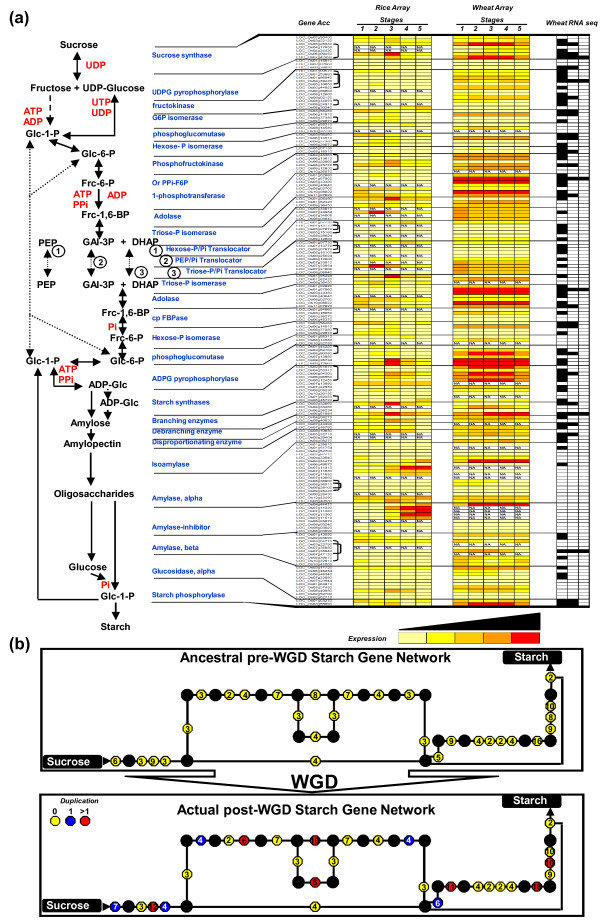
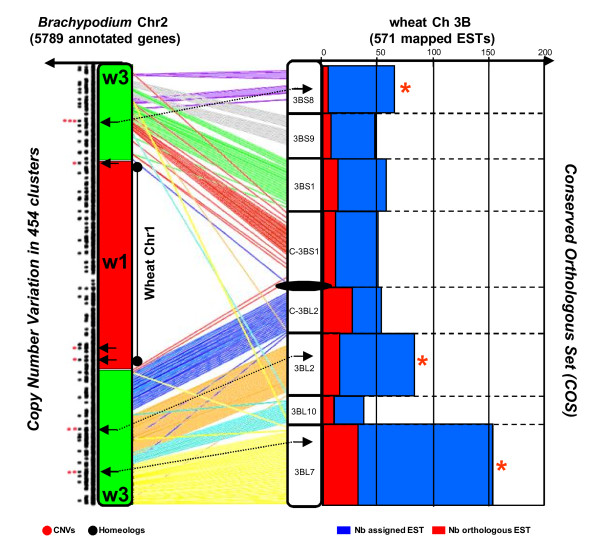
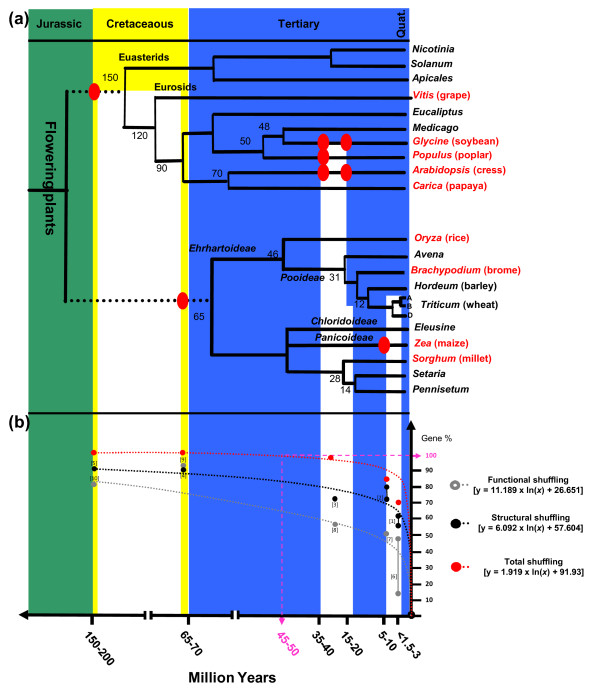
Results: We performed an RNA sequencing-based inference of the grain filling gene network in bread wheat and identified a set of 37,695 non-redundant sequence clusters, which is an unprecedented resolution corresponding to an estimated half of the wheat genome unigene repertoire. Using the Brachypodium distachyon genome as a reference for the Triticeae, we classified gene clusters into orthologous, paralogous, and homoeologous relationships. Based on this wheat gene evolutionary classification, older duplicated copies (dating back 50 to 70 million years) exhibit more than 80% gene loss and expression divergence while recent duplicates (dating back 1.5 to 3 million years) show only 54% gene loss and 36 to 49% expression divergence.
Conclusions: We suggest that structural shuffling due to duplicated gene loss is a rapid process, whereas functional shuffling due to neo- and/or subfunctionalization of duplicates is a longer process, and that both shuffling mechanisms drive functional redundancy erosion. We conclude that, as a result of these mechanisms, half the gene duplicates in plants are structurally and functionally altered within 10 million years of evolution, and the diploidization process is completed after 45 to 50 million years following polyploidization.
Figures
References
-
- Ohno S. Evolution by Gene Duplication. Berlin: Springer-Verlag; 1970. p. 160.
-
- Salse J, Abrouk M, Bolot S, Guilhot N, Courcelle E, Faraut T, Waugh R, Close TJ, Messing J, Feuillet C. Reconstruction of monocotelydoneous proto-chromosomes reveals faster evolution in plants than in animals. Proc Natl Acad Sci USA. 2009;106:14908–14913. doi: 10.1073/pnas.0902350106. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
